 Collagen vascular diseases are disorders of unknown etiology, with features of autoimmunity, in which the immune system loses the capacity to distinguish self from non-self. This loss of immune tolerance can result in varied and protean clinical manifestations, ranging from mild constitutional symptoms to major organ failure. Skin changes may be the initial manifestation of, or have pathognomonic features, for specific collagen vascular diseases, and are often invaluable to diagnosis. In this chapter we focus on identifying features of collagen vascular diseases, with an emphasis on cutaneous findings and emergent complications.
Collagen vascular diseases are disorders of unknown etiology, with features of autoimmunity, in which the immune system loses the capacity to distinguish self from non-self. This loss of immune tolerance can result in varied and protean clinical manifestations, ranging from mild constitutional symptoms to major organ failure. Skin changes may be the initial manifestation of, or have pathognomonic features, for specific collagen vascular diseases, and are often invaluable to diagnosis. In this chapter we focus on identifying features of collagen vascular diseases, with an emphasis on cutaneous findings and emergent complications.
I. Lupus Erythematosus
Lupus erythematosus is a broad term used to describe a spectrum of clinical entities ranging from localized cutaneous manifestations to potentially life-threatening, multi-organ systemic involvement, caused by pathogenic autoantibodies, leading to immune complex formation in target tissues.
Systemic lupus erythemtosus (SLE) is diagnosed clinically, as proposed by the American College of Rheumatology (ACR) criteria. Patients must fulfill 4 of the 11 criteria (1, 2) (Table 11.1).
Constitutional symptoms including fatigue, weight loss, and fever are common during active lupus. Fatigue may persist even when lupus is ‘quiescent’ (4, 5).
Arthritis, presenting as joint swelling and pain, and arthralgias, or joint pain without synovitis, are one of the most common, and early features of SLE, and are often present at sometime during the disease course (Table 11.2), specifically with a lupus flare (6).
Hematologic manifestations are also common, including leukopenia (46%), anemia (42%), and thrombocytopenia (7-30%) (3, 7). When leukopenia is secondary to active lupus, the WBC rarely decreases to
<1500 and the bone marrow is usually normal, with a greater drop in neutrophil count than lymphocyte count, though isolated lymphocytopenia is a more frequent finding. Anemia is frequent and often multifactorial, usually secondary to chronic disease or iron deficiency (8, 9).
Emergent Complications
Occasionally SLE patients can have severe life-threatening thrombocytopenia in the setting of a lupus flare, which can be refractory even to aggressive therapy. Thrombocytopenia may be part of the antiphospholipid syndrome or thrombotic thrombocytopenic purpura (TTP), which includes microangiopathic hemolytic anemia (suggested by schistocytes on peripheral smear), CNS deficits, renal failure and fever (10).
cardiopulmonary involvement is not infrequent. Pleuritic chest pain occurs in up to 50% of lupus patients at some time during their disease (11). Pericarditis is another common cause of chest pain in SLE patients, occurring ultimately in 19-48% of patients (12). Tamponade and myocardial disease is rare (13). Clinicians should consider pulmonary embolism as the cause of cardiopulmonary symptoms in patients with lupus anticoagulant or anticardiolipin antibodies (14). Pulmonary hemorrhage is a rare, yet life-threatening emergency in SLE, presenting with marked hypoxia along with abnormal chest imaging (15). This complication is usually the result of either pneumonitis or pulmonary embolus, and is associated with high mortality in the range of 50% to 90% (16). Lupus patients are at higher risk for coronary artery disease, and myocardial infarction is now the most common cause of death in SLE patients in the Western world (17, 18).
kidney involvement is a major cause of morbidity and hospital admissions. The most common manifestation of lupus nephritis is proteinuria (19, 20). Nephritic or nephrotic syndrome can be observed in 30% of patients. Rapidly progressive glomerulonephritis occurs in less than 5% of patients with SLE (21).
Neuropsychiatric manifestations are seen in up to 60% of patients. Symptoms include seizures, personality changes, and cerebrovascular accidents, and are a major cause of morbidity (22, 23, 24).
CUTANEOUS LUPUS
Cutaneous lupus is seen in 59-85% of SLE patients (25), and can be classified as LE-specific skin disease and LE-non-specific skin disease. LE-specific skin disease has characteristic histopathologic changes, including epidermal atrophy, hyperkeratosis, liquefactive degeneration of the basal epidermis and mononuclear cell infiltrate (26). The three main categories of LE-specific cutaneous disease are acute cutaneous (ACLE), subacute cutaneous (SCLE), and chronic cutaneous (CCLE).
LE non-specific lesions such as vasculitis and alopecia, do not have the characteristic LE histopathological changes, and can found in other medical conditions besides lupus.
It is important to note that not all cutaneous lupus patients have systemic lupus. Of patients with discoid lupus erythematosus (DLE) lesions, only 5-10% will go on to develop SLE. Conversely, 25% of patients with systemic lupus develop DLE lesions at some point in their disease course. Of patients with SCLE, about 10-15% develop SLE. Presence of acute cutaneous lupus lesions usually indicates underlying SLE (27, 28).
LE specific skin disease
Acute Cutaneous Lupus Erythematosus (ACLE): ACLE can present in localized or generalized distributions and often occurs in the setting of acutely flaring SLE (29). The most common form of ACLE is localized symmetric, confluent erythema and edema over the malar region and the bridge of the nose, sparing the nasolabial folds, also known as the “butterfly” rash (Fig 11.1). The malar butterfly rash is present at time of SLE diagnosis in 20-60% of patients (25, 30). The eruption fluctuates with disease activity and can be differentiated from acne rosacea or seborrheic dermatitis by its lack of papules and pustules, and by its distribution. Painless oral and/or nasal mucosal ulcerations (Fig 11.2) frequently accompany lesions of ACLE (31).
Generalized ACLE presents similarly to a viral exanthem or drug eruption, as a widespread morbilliform eruption, with diffuse edema and erythema of the face, upper trunk, or extremities (Fig 11.3). The lesions develop rapidly and last for hours to days. There may be associated edema and erythema of the hands over the dorsal and interphalangeal areas, with pathognomonic sparing of the skin overlying the knuckles, in contrast to Gottron’s papules of dermatomyositis. Generalized ACLE can affect up to a third of patients with SLE (29). Severe forms of generalized ACLE can present with vesiculobullous skin changes, due to widespread apoptosis of epidermal keratinocytes, clinically resembling toxic epidermal necrolysis (TEN). However, unlike TEN, bullous ACLE is photosensitive, occurring predominantly over sun-exposed areas, has a more insidious onset, and may or may not involve mucosa (32). Outside of the skin, there can be bullous LE involvement of ocular, esophageal or laryngeal tissue, potentially leading to corneal blindness and laryngoesophageal stenosis. Histopathology and immunofluorescence studies are necessary to delineate bullous SLE from other blistering diseases in the lupus patient (29). A patient presenting with vesiculobullous lesions should be treated emergently regardless of etiology (see vesiculobullous diseases and drug eruptions chapters).
Subacute Cutaneous Lupus Erythematosus (SCLE): SCLE presents as erythematous macules and/or papules that evolve into hyperkeratotic, papulosquamous annular plaques often occurring often over the neck, shoulders, upper extremities, and trunk (Fig 11.4), resembling psoriasis (33). About 70% of SCLE patients have positive anti-Ro/SSA antibodies. SCLE patients are, in general, more likely to progress to SLE than those with chronic cutaneous lupus (34). Severe systemic manifestations such as systemic vasculitis, renal disease, and CNS occur in less than 10% of patients with SCLE (29). Risk factors for the development of SLE in a patient with SCLE lesions include papulosquamous SCLE, leukopenia, and antinuclear antibody (ANA) titer greater than 1:640 (35). SCLE is drug-induced in 12% of cases. It resembles idiopathic SCLE clinically and histopathologically, but resolves when the offending drug, most often an antihypertensive or antifungal agent, is withdrawn (33,34).
Chronic Cutaneous LE (CCLE): The most common form of CCLE is discoid lupus, which begins as red-purple macules and papules that evolve into coin-shaped, erythematous plaques with scale extending into the openings of dilated hair follicles (Fig 11.5). Central scarring, telangiectasias, and hypopigmentation are the post-inflammatory hallmarks of these lesions, most striking on darker skinned patients, and can be very disfiguring. DLE is commonly found on the head and neck and extensor aspects of the arms, similar to ACLE, but is neither edematous, nor transient. There can be mucosal DLE involvement of the oral and genital mucosa, and the conjunctiva (36). The small percentage of DLE patients that progress to develop SLE are more likely to do so within the first five years of DLE diagnosis, and usually have generalized DLE distribution (lesions both above and below the neck) (37). SLE patients with DLE skin lesions tend to have less severe systemic lupus activity.
Other variants of CCLE are lupus panniculitis, lupus tumidus, and chilblains lupus. Lupus panniculitis (or lupus profundus) presents as firm, depressed nodules, extending into the deep dermis, and is seen in 1-3% of CCLE patients (Fig 11.6) (38). About 10-40% of those with LE panniculitis have mild manifestations of SLE (39). Lupus tumidus presents as erythematous, urticarial plaques with minimal surface change and no follicular plugging (Fig 11.7) (40). Lupus tumidus patients rarely have clinically evident SLE and are usually ANA negative. Chilblain lupus, a rare form of DLE, presents as violaceous plaques and papules on the tips of fingers and toes, exacerbated by cold exposure (Fig 11.8). 20% of chilblain lupus patients go on to develop SLE (41).
LE Non-Specific Skin Disease
Cutaneous vasculitis is reported to occur in 11% of SLE patients (Fig 11.9) (43). Palpable purpura is most common, often in the lower extremities, although a generalized or acral distribution can also be observed (29). Less dependent areas of the skin can exhibit urticarial vasculitis. Additionally, fingertip gangrene, splinter hemorrhages, periungual telangiectasia, and purpuric lesions of the palm can be seen. Antiphospholipid syndrome or infective endocarditis should be kept in the differential when evaluating these patients (43). Cutaneous vasculitis may be a predictor of the future development of LE nephritis (44).
Alopecia is common in lupus patients (45). Telogen effluvium can occur with systemic flares, and tends to resolve with lupus treatment. Discoid lupus can cause a scarring alopecia (29). Non-scarring alopecia is seen in 24% of patients with SLE (30).
Cutaneous mucinosis is an unusual non-specific presentation of SLE, presenting as asymptomatic papules and nodules on the trunk and arms, with increased mucin deposition in the dermis on histology. In one study, this occurred in 1.5% of SLE patients. Another study found that 80% of patients with this presentation had underlying SLE, though it can also be seen in other connective tissue diseases, such as dermatomyositis and scleroderma (46, 47)
Other nonspecific cutaneous findings include Senear-Usher syndrome, or pemphigus erythematosus, which manifests as pemphigus foliaceous-like lesions in a seborrheic distribution in the setting of LE, and Rowell syndrome, in which erythema multiforme-like lesions are seen in patients with LE (31, 48).
Laboratory findings
Whereas ANA may or may not be positive in isolated cutaneous lupus patients, ANA >1:160 is present in virtually all SLE patients (27). Anti-double-stranded DNA antibodies (dsDNA), seen in 70% of patients, and anti-Smith antibodies (anti-Sm), seen in 25% of patients, are both highly specific for SLE (25). Along with falling complement levels, rising anti-dsDNA titers are markers for an acute or impending SLE flare. With a lupus flare, the CBC may reveal leukopenia, hemolytic anemia, and/or thrombocytopenia. (7, 25, 49).
All lupus patients should be monitored for proteinuria and active urinary sediment, such as cellular casts and hematuria, which are indicative of lupus nephritis (50). As ACLE often occurs in the setting of active systemic lupus, these patients should have thorough evaluation for underlying disease activity including a history and physical examination, complete blood count, comprehensive metabolic panel, urinalysis, complement levels and autoantibody profile.
Treatment
Antimalarials, particularly hydroxychloroquine, are used almost universally in SLE patients, except in those with intolerance or G6PD enzyme deficiency (51). Hydroxychloroquine is safe in pregnancy and lactation, prevents and decreases severity of lupus flares, and has the added benefit of increased survival, decreased thrombosis and improved lipid levels in patients who take the medication compliantly (52, 53). Patients on antimalarials should be screened for maculopathy and neuromyopathy. Another side effect is increased blue-gray skin pigmentation with chronic hydroxychloroquine ingestion (25, 54, 55).
Lupus flares are treated according to severity and risk to major organ function, with glucocorticoids being the cornerstone of treatment. Mild to moderate flares, arthritis, and serositis can be treated with low dose prednisone 10-20 mg per day, while moderate to severe flares require higher doses. Steroids are tapered according to patient response (56). For patients who flare with steroid tapering, or have recurrent lupus flares, or severe organ manifestations, maintenance immunosuppression with azathioprine or mycophenolate mofetil (25, 57), amongst others, should be considered. For patients who fail these standard therapies, the newest FDA-approved medication for moderate to severe SLE, IV Belimumab, a B-lymphocyte stimulator-specific inhibitor, may be considered.
Most hospitalized patients and those with severe flares are treated with the equivalent of prednisone 1 mg/kg/day, or pulse doses of IV methylprednisolone: one gram/day for three days, particularly for cases of rapidly progressive lupus nephritis or life/organ-threatening manifestations. For patients with life-threatening organ manifestations such as severe lupus nephritis or neuropsychiatric disease, six monthly doses of IV cyclophosphamide may be added to high dose steroids. Due to concern over cyclophosphamide toxicity, azathioprine can be substituted for mild forms of lupus nephritis, and mycophenolate mofetil can be effective in induction treatment for even moderate to severe proliferative lupus nephritis, especially in African Americans and Hispanics (58). Intravenous immunoglobulin (IVIG) has shown benefit in autoimmune hemolytic anemia and thrombocytopenia, and can be used for other severe lupus manifestations, without increasing risk for secondary infection (59).
Because cutaneous lupus is highly photosensitive, sun protection is paramount, including the use of sunblocks and sunscreens, as well as hats and sun-protective clothing (60). Initial therapy usually includes topical glucocorticoids, and/or intralesional steroid injection. Antimalarials such as hydroxychloroquine with or without quinacrine are added first as systemic therapy or in combination if topical or local therapy is inadequate (61). Patients with extensive skin involvement may require oral corticosteroids, and if needed, azathioprine, methotrexate, mycophenolate mofetil, dapsone (particularly for bullous lesions), or thalidomide can be added (25, 29).
NEONATAL LUPUS
One third of SLE patients have antibodies to the SSA/Ro and SSB/La antigens. Neonatal lupus (NLE) is caused by placental transfer of either SSA/Ro, or SSB/La antibodies and/or U1-RNP to the fetus. (62). The risk of a baby being born with NLE to an SSA/Ro+ mother is roughly 20%, and increasing to 25% in women who have previously given birth to an affected child. An SCLE-like annular rash occurs in 20% of babies born to antibody positive mothers. Unlike classic SCLE, neonatal lupus can scar (Fig 10).Congenital heart block occurs in 1-3% neonatal lupus, thought to result from anti-Ro/La antibodies attacking cardiac conduction tissue (63). Congenital heart block can be diagnosed by fetal echocardiography, usually starting at gestational week 18, and is treated with IV steroids administered to the mother (62, 64). Mortality from congenital heart block is roughly 20-30%. There is also a risk of liver failure secondary to hepatobiliary disease and thrombocytopenia. Most deaths occur in utero or within the first three months after birth (62, 63, 65. NLE without cardiac involvement usually spontaneously resolves within 2-6 months, though during that period, maternal antibodies persist and can precipitate new skin lesions upon exposure to UV light (66) .
ANTIPHOSPHOLIPID SYNDROME
Antiphospholipid syndrome (APS) is defined by persistent serum antiphospholipid antibodies [anticardiolipin (ACL), lupus anticoagulant (LAC), or anti-2-glycoprotein I antibodies (anti-2GPI)], along with vascular thromboses and/or pregnancy morbidity (67). Primary disease occurs in 1% of the population (11). Patients with concomitant autoimmune disease such as lupus, are labeled as ‘secondary APS’. Of lupus patients with antiphospholipid antibodies, 10-15% have clinical manifestations of APS (68, 69).
Up to 50% of APS patients have nonbacterial cardiac valvular thickening or vegetations, known as marantic or Libman-Sacks endocarditis. Though severe cardiac manifestations requiring valvular replacement are rare, dermatologic manifestations are common. Livedo reticularis is seen in 22-35% of SLE patients (43), particularly those with antiphospholipid syndrome (Fig 11.11). Other nonspecific APS skin manifestations include atrophie blanche, painful extremity ulcers that heal as a white atrophic scar, (Fig 11.12), and embolic phenomenon leading to acral purpura and gangrene (70).
Emergent complications
Thrombosis may occur in large, medium, and small vessels of the venous and arterial circulation. Venous thromboses are most common in the legs, but have been reported in varied sites not limited to the renal, mesenteric, axillary and sagittal veins. Arterial events can be the result of either primary thrombosis or from emboli originating from valvular vegetations. Transient ischemic attacks are the most frequent manifestation of arterial thrombosis in APS (67, 71).
APS is a significant cause of recurrent pregnancy loss in lupus patients. Pregnancy morbidity can manifest as unexplained consecutive spontaneous abortions before the 10th week of gestation, death of a normal fetus beyond the 10th week of gestation, or premature birth of a normal neonate prior to 34th week of gestation due to eclampsia, severe preeclampsia or placental insufficiency. Those occurring after 10 weeks of gestation are most associated with aPL. Patients with a history of previous fetal loss and high titer IgG aCL are at 80% risk for loss of their current pregnancy. Other obstetric manifestations of APS include premature delivery, oligohydramnios, and intrauterine growth restriction (72, 73) .
Catastrophic antiphospholipid syndrome (CAPS) is defined as positive antiphospholipid antibodies along with three or more organ thromboses occurring over a short period of time. Occlusion of small vessels, or thrombotic microangiopathy, is characteristic. CAPS should be distinguished from thrombotic thrombocytopenic purpura and disseminated intravascular coagulation. CAPS is rare, occurring in only 0.8% of patients with APS, but is significant due to high mortality rate (74).
Laboratory findings
APS laboratory criteria require at least one of the three following tests, present on two or more occasions in a period of twelve weeks: lupus anticoagulant, IgG or IgM anticardiolipin antibodies in medium-high titers, or IgG, IgM, or anti-2-GPI antibodies above the 99th percentile (67, 76, 77, 78).
False positive tests for syphilis often indicate the presence of underlying antiphospholipid antibodies. Besides being associated with autoimmune disease, APS antibodies and can be drug-induced, or infection related (78). Medications associated with aPL are hydralazine, procainamide, and phenytoin. Bacterial infections, including syphilis, Lyme disease, post-streptococcal rheumatic fever, and viral infections, such as hepatitis A, B, C, mumps, HIV, varicella zoster, EBV, and parvovirus can all result in IgM aCL in the serum (79).
Other lab abnormalities seen in APS are thrombocytopenia (typically mild), and less commonly, Coombs-positive hemolytic anemia. Antiphospholipid antibodies with thrombocytopenia and autoimmune hemolytic anemia is known as Evans syndrome (80).
Treatment
Patients with APS and history of thrombosis require lifelong anticoagulation at an INR of 2-3 for venous events. There is controversy over whether the INR should be kept at a higher level for arterial thrombosis (81). Some studies suggest that APL antibody positive SLE patients may benefit from prophylactic aspirin therapy (82). Severe cases of APS associated thrombocytopenia are treated with steroids, IVIG or splenectomy if resistant (83, 84).
Catastrophic antiphospholipid syndrome patients are in general very ill, and require anticoagulation, high dose steroids and plasma exchange (75).
Women with APS and recurrent miscarriages are treated with low dose aspirin along with either heparin or low molecular weight heparin (LMWH) during subsequent pregnancies (85). Anticoagulation is continued for six weeks after delivery (87). Intravenous immunoglobulin with or without steroids is also being evaluated in clinical trials for use in preventing pregnancy losses in women with APL. Pregnant APS patients are at risk for and should be monitored for preeclampsia. The fetus should be monitored for placental insufficiency the last weeks of the third trimester (87).
II. Dermatomyositis
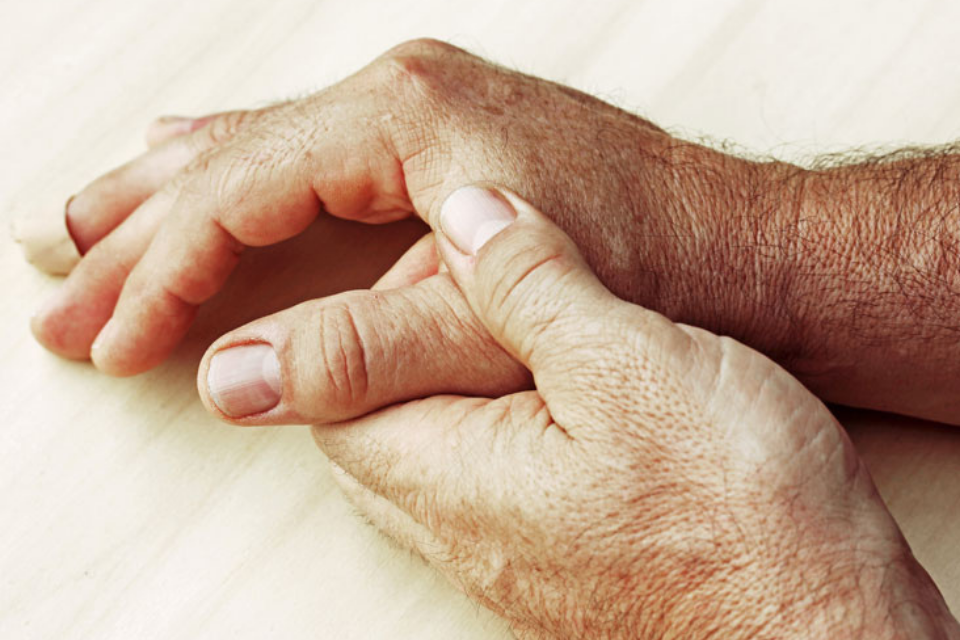
Dermatomyositis (DM) is an inflammatory muscle disease associated with characteristic skin manifestations. The cutaneous findings pathognomonic for DM are: Gottron’s papules, violaceous or pink papules of the dorsal metacarpal and interphalangeal joints (Fig 11.13), and Gottron’s sign, macular erythema over extensor surfaces such as the elbows and knees (Fig 11.14). Lesions resemble cutaneous lupus in that they are sensitive to UV light and have similar histopathologic findings. Also frequently occuring are the heliotrope rash, presenting as periorbital violaceous erythema (Fig 11.15), and erythema extending over the shoulders (the “shawl sign”), or the anterior chest(the “V-sign”), which can later progress to poikiloderma. Nail fold changes are also common, including periungual erythema and telangiectasias (88).
DM is most commonly diagnosed using the Bohan and Peter Criteria. According to the criteria, a patient has definite DM if presenting with a typical DM skin rash, and any three of the following: symmetric proximal muscle weakness, elevated skeletal muscle enzymes, characteristic electromyogram, or myositis on muscle biopsy, (89). Amyopathic DM is defined as a characteristic DM rash, without clinical, laboratory or histologic evidence muscle involvement for at least 2 years following initial presentation (90).
Emergent Complications
Interstitial lung disease (ILD) is the most frequent cause of morbidity and mortality in DM (91, 92), and is usually associated with the presence of anti-Jo1 antibody. An important subset of myositis is the antisynthetase syndrome: inflammatory myositis associated with presence of anti Jo-1 antibody, along with ILD, fever, Raynaud’s phenomenon, arthritis, and “mechanics hands” (rough erythema on the sides of the fingers and palm) (93, 94). Of amyopathic DM patients, up to 25% have ILD and must be screened with annual pulmonary function tests (95). The natural course of ILD is gradual and progressive, though acute deteriorations can occur (94, 96). Besides ILD, dyspnea can be due to weakness of the diaphragm and thoracic muscles, or from myositis of the striated esophageal muscles, which can lead to aspiration pneumonia.
Cardiac myositis, another complication of DM, is usually subclinical, presenting with mild rhythm disturbances. Congestive heart failure from myocarditis or fibrosis, is rare. Myocardial involvement, pharyngeal weakness, malignancy, ILD, and delayed initiation of steroids are associated with poor survival (97, 98).
Several population-based studies have documented a higher-than-expected risk of malignancy in patients with adult-onset DM (99, 100, 101). Risk of malignancy increases with age, male gender, and elevated serum creatine kinase levels (102). The most commonly associated malignancies are ovarian, breast, lung, pancreatic, stomach, colorectal cancer, and non-Hodgkin lymphoma (103). Risk of malignancy appears to approach that of the general population if no malignancy is discovered within the first two years of diagnosis. (104).
DM can also occur in the pediatric population, with a similar rash as adult DM, except with a more frequent incidence of soft tissue calcinosis and cutaneous or gastrointestinal ulceration and vasculitis (105). Unlike adult dermatomyositis, malignancy is rarely associated with juvenile dermatomyositis. Up to 15-24% of adult individuals with dermatomyositis are diagnosed with malignancy either at time of diagnosis of DM or soon thereafter, in contrast to only 1% of pediatric patients with juvenile dermatomyositis (106).
Laboratory findings
Evidence of myositis includes elevated creatine kinase (CK), aldolase, and liver function tests. CK is most often used for monitoring of disease activity and response to therapy (107).
Treatment
Initial pharmacologic therapy for DM consists of high-dose glucocorticoids, tapered once muscle enzymes normalize (108). Steroid sparing agents may be added, most commonly azathioprine and/or methotrexate, particularly in patients with major organ involvement, delayed diagnosis or treatment, or severe myositis. For treatment resistant myositis, IVIG, mycophenolate mofetil, and rituximab may provide benefit (109, 110).
The rash of dermatomyositis often responds to myositis treatment, along with sun avoidance, topical steroids, and hydroxychloroquine. Malignancy associated myositis and DM rash are generally refractory to steroids, resolving only with treatment of the underlying cancer (103, 111)
III. Systemic sclerosis
Systemic sclerosis (SSc), also known as scleroderma, is characterized by microvascular injury and excessive collagen deposition and fibrosis of the skin and internal organs. The extent of skin sclerosis determines whether the patient has a diffuse or limited systemic sclerosis. Each subset has unique serologic markers and variable associations with underlying organ manifestations.
Diffuse systemic sclerosis (dSSc) is defined by skin changes extending from the fingers and toes proximally to the elbow and knees, often involving the chest and abdominal wall. With progressive skin tightening and fibrosis, certain areas of the skin may become hypo- or hyperpigmented. Oftent, pigment is maintained around the hair follicle, leading to a “salt and pepper” appearance of the skin (Fig 11.16). Disease presentation is usually abrupt, and symmetric hand edema and Raynaud’s phenomenon are closely related to the onset of sclerosis. While skin sclerosis is the most severe in the first eighteen months of disease and can subsequently improve, disease involvement of internal organs continues to progress with time (112, 113).
Limited systemic sclerosis (lSSc) presents with a more indolent course than dSSc. Patients often have a longstanding preexisting history of Raynaud’s phenomenon, later presenting with skin thickening and fibrosis distal to the elbows and knees. Both diffuse and limited SSc can have taut facial skin yielding a mask-like appearance, with a small oral aperture (Fig 11.17). Besides Raynaud’s, other lSSc features include subcutaneous calcinosis, gastrointestinal reflux and esophageal dysmotility, sclerodactyly, and telangiectasias, also known as CREST syndrome. One percent of cases can develop internal organ involvement without characteristic skin changes (114). Limited SSc is associated with a higher risk of pulmonary hypertension, but overall prolonged survival when compared to dSSc (115, 116).
Scleroderma can overlap with other autoimmune diseases, including the inflammatory myopathies, SLE, and Sjögren’s syndrome.
Emergent complications
Until the development of ACE inhibitors, scleroderma renal crisis (SRC) was the most serious and lethal complication of SSc (117). SRC, arising from vessel narrowing and ischemic kidney disease, presents almost exclusively during the first 5 years of diffuse SSc, as new-onset hypertension, acute renal failure, and microangiopathic hemolytic anemia (118).
Nowadays, most disease-related SSc mortality is from cardiopulmonary involvement: interstitial lung disease (ILD), subsequent pulmonary fibrosis, and pulmonary artery hypertension (PAH). Patients are monitored for lung disease with pulmonary function tests and/or imaging (119). The greatest decline in lung function from ILD occurs in the first 4-6 years of scleroderma onset (120, 121).
Pulmonary arterial hypertension (PAH) can occur in any subset of scleroderma, but as mentioned earlier, is more frequent in limited SSc, usually occurring years after diagnosis. Patients are monitored for PAH by echocardiography, and diagnosis is confirmed by right heart catheterization (122).
Besides PAH, other cardiac features include pericardial effusions, arrhythmias and diastolic dysfunction. Myocardial ischemia, fibrosis, and myocarditis have been noted as well. (123, 124).
Empiric treatment with a proton pump inhibitor is recommended to prevent SSc gastrointestinal complications, including dysmotility, reflux, and stricture formation. (125). SSc patients may have gastrointestinal blood loss from mucosal telangiectasias, such as gastric antral vascular ectasia (GAVE), described endoscopically as “watermelon stomach”. (126). There is an association between primary biliary cirrhosis (PBC), which often has an associated anticentromere antibody, and limited scleroderma. Without ursodiol treatment, there can be progression to secondary cirrhosis. (127). Intestinal fibrosis can yield dysmotility, giving rise to bacterial overgrowth, abdominal pain and bloating.
Finally, Raynaud’s phenomenon of scleroderma is typically severe and resistant to treatment, leading to critical digital ischemia, ulceration, gangrene, and ultimately, auto-amputation (Fig 18). Ischemic pain and decreased capillary refill with or without signs of visible tissue damage is a sign of ongoing ischemia and requires immediate treatment. Care should be taken to not mistake either digital calcinosis, or sores from microtrauma of taut atrophic skin covering joints, for digital ulcerations (128).
Laboratory findings
Most SSc patients will have a positive ANA. A negative ANA should alert the clinician to consider other diagnoses such as nephrogenic systemic fibrosis, scleromyxedema, scleredema, graft versus host disease (GVHD), and toxin-mediated sclerotic syndromes (129). The anti-centromere antibody is almost exclusively seen in lSSc. Anti-Scl antibody is more characteristic of dSSc, especially those with ILD, and is found in 20-30% of patients. Anti-RNA polymerase I/III antibodies are linked to scleroderma renal crisis (130, 131).
Treatment
Most treatment is directed towards a specific organ and/or symptom. For Raynaud’s, vasodilatation and increased blood flow to the digits is the goal of treatment and can include any combination of calcium channel blockers, angiotensin receptor blockers, nitropaste, or even sildenafil and/or bosentan, if resistant secondary ulcers are present.
Proton pump inhibitors are beneficial for esophageal dysmotility, strictures and esophagitis. Promotility agents such as metoclopramide alternating with antibiotics can alleviate symptoms of bacterial overgrowth.
Many immunosuppressant agents have been used as disease modifying treatments in systemic sclerosis, including mycophenolate mofetil, azathioprine and methotrexate, but only cyclophosphamide has been shown in randomized controlled studies to be of benefit in SSc ILD, albeit modestly. (119, 132). Corticosteroids play a minimal role in the management of scleroderma, and are used mainly for the treatment of associated inflammatory arthritis (133).
Depending on the severity of pulmonary arterial hypertension (PAH), oral endothelin receptor antagonists, phosphodiesterase inhibitors, inhaled iloprost, or prostacyclin can be used (134, 135).
IV. Relapsing Polychondritis
Relapsing polychrondritis (RP) is characterized by painful inflammation of cartilage throughout the body. Autoantibodies and cell-mediated immunity towards extracellular matrix components, including collagen types II, IX, and XI, have been implicated in the pathogenesis. Episodes last days to weeks and can resolve spontaneously. The classic finding in RP is acute uni- or bilateral auricular chondritis (Fig 11.19). Recurrent attacks of auricular chondritis can result in the floppy “cauliflower ear,” and attacks of nasal cartilage can yield a depressed “saddle nose” deformity. Nasal involvement is common, presenting as a tender, warm, and red nasal bridge. Up to 12% of patients may present with non-specific dermatologic findings, including erythema nodosum, purpura, livedo reticularis, and distal necrosis. (136,137).
Emergent complications
Involvement of the cartilaginous rings of the respiratory tract is a major cause of mortality, and can occur in over 50% of patients (138). Though there is little correlation between severity of clinical symptoms and the extent of airway involvement, clinicians should consider laryngotracheal involvement in any RP patient with respiratory complaints, including hoarseness, non-productive cough, wheezing, dyspnea, or stridor. Flow-volume loop studies should be obtained for all patients diagnosed with RP and be repeated every 6-12 months to evaluate for sub-acute airway strictures and narrowing. During an acute attack, CT scan may show edema and thickening of the trachea. Recurrent pneumonias or bronchiectasis may suggest a collapsed bronchus (139, 140).
Less than 10% of cases have cardiovascular involvement. Aortic regurgitation secondary to aortic root dilatation can occur (141). Other manifestations include mitral regurgitation and conduction abnormalities (142).
Renal dysfunction is seen in up to one quarter of patients, manifesting as IgA nephropathy, tubointerstitial nephritis, or segmental necrotizing crescentic glomerulonephritis (143). Renal involvement is associated with extra-renal vasculitis, arthritis, and overall worse prognosis (144).
Ocular inflammation is found in up to 60%. Episcleritis and scleritis are most frequent, followed by keratoconjunctivitis, sicca, uveitis, and ulcerative keratitis. Although rare, blindness can result from corneal perforation, retinal vasculitis, and optic neuritis (145).
There is an association between myelodysplasia and RP, especially in patients who are male, have dermatologic manifestations and an older age at onset of disease. (135, 146).
Laboratory findings
There is no specific screening autoantibody for RP. Anti-collagen antibody tests are not always readily available, and may not be positive in all cases. A positive ANA is seen in 22 to 66 percent of patients. In atypical cases, biopsy of involved cartilage may be needed to confirm diagnosis (135).
Treatment
Initial treatment of relapsing polychondritis consists of low dose prednisone for mild auricular or nasal chondritis, or arthritis, while more serious manifestations such as laryngeotracheal or ocular symptoms require higher doses. For steroid dependent or refractory cases, anecdotal evidence suggests dapsone and colchicine, or if severe, cyclophosphamide, azathioprine, cyclosporine, or mycophenolate mofetil may be helpful. Biologics such as infliximab and the IL-1 receptor antagonist, Anakinra, have been used as well. Localized steroid therapy can be used in the form of eye drops, inhaled steroids, or intra-articular or intralesional injections (147, 148, 149, 150).
V. Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a chronic inflammatory polyarthritis, with a predilection for the small joints of the hands, feet and wrists. The arthritis is commonly symmetric, affecting upper and lower extremity joints, with associated morning stiffness lasting for hours. If left untreated, or suboptimally treated, chronic untreated joint inflammation can progress to a secondary osteoarthritis and deforming arthritis with bone erosions on imaging. Among the more common cutaneous findings in RA are rheumatoid nodules, seen in 20-30% of patients. Rheumatoid nodules are painless, firm, flesh-colored subcutaneous nodules, occurring over extensor surfaces, believed to occur from small vessel vasculitis with fibrinoid necrosis. Rheumatoid nodules portend a more severe disease course and higher risk of developing extraarticular disease (150). Rheumatoid nodules are more common in males and rheumatoid factor (RF) positive patients (151). Neutrophilic dermatoses can also present in patients with RA, including pyoderma gangrenosum, Sweet’s syndrome, rheumatoid neutrophilic dermatitis, erythema elevatum diutinum, and palisading neutrophilic and granulomatous dermatitis (152, 153).
Bywater’s lesions are small brown to purpuric painless lesions on the nail fold, nail edge or digital pulp that represent nail fold infarctions, and do not require intense immunosuppression (154, 155).
Emergent Complications
RA is associated with exudative pleural effusions with low fluid glucose levels that can be mistaken for empyema. Patients with cutaneous rheumatoid nodules can have associated pulmonary nodules, which are usually asymptomatic, but can rarely cavitate and cause bronchopleural fistulas or pneumothorax. Symptomatic interstitial lung disease is reported in up to 10% of RA population studies. Some forms of RA-associated ILD such as cryptogenic obstructive pneumonia are very steroid responsive, whereas usual interstitial pneumonitis is progressive despite treatment (156).
Cervical spine involvement is frequent in patients with severe RA. Patients complaining of neck pain or upper extremity paresthesias should alert the clinician to possible cervical spinal cord compression from atlantoaxial subluxation. Other symptoms include sensation of the ‘head falling off’, weakness, incontinence, and positive Babinski reflex. Atlantoaxial subluxation occurs as a result of a lax C1 transverse ligament or from erosion of the odontoid process. Diagnosis is confirmed with a lateral flexion-extension x-ray, in which the space between the C1 anterior arch and C2 odontoid process is greater than 5 mm. Basilar invagination can also occur, from upward migration of the odontoid process into the foramen magnum. Both processes can lead to vertebral artery compression (157).
Active RA can be associated with episcleritis, scleritis, and peripheral ulcerative keratitis. Scleritis, when untreated, can progress from eye redness and pain to necrotizing scleritis, which is associated with underlying vasculitis, vision loss, and high mortality (169).
Felty’s syndrome, consisting of RA, leukopenia and splenomegaly, occurs in one percent of RA patients, mainly those with long standing, seropositive, erosive and deforming RA (158). Patients with Felty’s syndrome frequently have lower extremity ulcers as well as an increased risk of melanoma, lymphomas, and leukemias (159). Leukopenia portends a 25% mortality risk from sepsis (160).
Rheumatoid vasculitis is uncommon (
<5%), and typically occurs in patients with long-standing, erosive RA with RF seropositivity and rheumatoid nodules. Cutaneous manifestations are digital gangrene and leg ulcers along the lateral malleolus or pretibial region, and nonspecific erythema, hemorrhagic blisters, livedo reticularis, erythema elevatum diutinum and atrophie blanche (161). Visceral involvement of different organ systems (eye, heart, lung, GI) and the peripheral nerves can occur (162, 163). Features include scleritis, pericarditis, myocardial infarction, neuropathy and acute abdomen. (164, 165, 175). Decreased survival has been noted in patients with a younger age at diagnosis, delayed diagnosis, abnormal urinary sediment, and hypergammaglobulinemia (167). Cardiac involvement, gangrene, bowel infarctions and mononeuritis multiplex also suggest poor prognosis (165, 166). As the pathogenesis is immune complex mediated, it has been suggested that direct immunofluorescence of skin biopsy may detect early or subclinical vasculitis in RA patients (168).
Laboratory findings
Rheumatoid factor is an autoantibody against IgG, seen in 80% of RA patients, but can also be seen in Sjögren’s syndrome, cryoglobulinemia and SLE amongst others diseases. Low titers may also be seen in the elderly, and patients with long standing infection. Anti-citrullinated peptide antibodies (anti-CCP antibody or ACPA) can be see in up to 85% of RA patients later in the course of the disease, and has higher specificity for RA than the RF does.
The 2010 ACR/EULAR classification incorporates joint involvement, serology, duration of symptoms and acute phase reactants for diagnosis. The acute phase reactants, erythrocyte sedimentation rate (ESR) and C reactive protein (CRP), are also used in monitoring disease activity. (173, 174)
Treatment
Once rheumatoid arthritis is diagnosed, aggressive therapy with disease-modifying antirheumatic drugs (DMARDs) should be initiated, in order to prevent further joint damage and subsequent permanent deformity.
Methotrexate is considered the anchor drug. If disease control is not achieved, other DMARDs such as leflunomide, or sulfasalazine with hydroxychloroquine can be added, or biologic agents can be employed. Adjunctive treatments for helping with pain and function include glucocorticoids, NSAIDs, and physical and occupational therapy.
Treatment of rheumatoid vasculitis requires high dose glucocorticoids with or without cyclophosphamide. Methotrexate is not only a DMARD, but is also used to improve Felty’s syndrome associated cytopenias. Splenectomy is indicated for recurrent infections or noncorrecting leukopenia, and may also improve leg ulcers. (174, 175, 176)
Scleritis requires topical or local steroids with or without systemic immunosuppressants. Hematologic findings such as anemia of chronic disease and thrombocytosis, tend to resolve as the RA comes under control with immunosuppression. Peripheral neuropathy from inflamed synovial entrapment will also improve with RA treatment. Atlantoaxial subluxation greater than 8 mm from cervical involvement usually requires surgical intervention. (169, 173, 177, 178, 179)
VI. Sjögren’s Syndrome
Sjögren’s syndrome (SS) is an autoimmune disorder characterized by lymphocytic infiltration of exocrine glands, primarily manifesting as xerophthalmia and xerostomia. Sjögren’s syndrome can be a primary disorder, or can be seen in association with other autoimmune diseases such as SLE and RA (179). The disease can manifest with a variety of cutaneous signs, the most common being xerosis. Cutaneous vasculitis, including crygolubulinemia, may suggest more serious systemic and extraglandular involvement, (180, 181).
Lymphocytic infiltration of various organs can yield interstitial pneumonitis, pericarditis, and interstitial nephritis. These features usually appear early in the disease and follow a benign course. Arthritis and arthralgias are seen in over half of patients. Primary biliary cirrhosis and celiac sprue have also been noted to be associated with SS (182, 183).
Emergent complications
There is a 10 to 40-fold increased risk of lymphoma in SS patients. The overall risk of developing lymphoma is 5% in patients with SS. Patients with SS also have a predilection for vasculitis, and in particular, type II cryoglobulinemic vasculitis is associated with lymphoma (most commonly non-Hodgkins type) both independently, as well as in those with SS. Other risk factors for lymphoma include C4 hypocomplementemia and hard enlargement of parotid or lacrimal glands (184). Neurologic complications include sensorimotor neuropathy, though cranial neuropathies, central demyelinating disease, and optic atrophy have also been described. Central nervous system vasculitis is uncommon (180, 185).
Three prognostic factors for adverse outcomes in primary SS are purpura, hypocomplementemia, and cryoglobulinemia, as identified in several prospective studies of SS pts. Such outcomes include systemic vasculitis, B cell lymphoma, and death (186, 187, 188)
Laboratory findings
The presence of SSA/Ro and SSB/La antibodies are part of the diagnostic criteria, and occur in high frequency, along with RF and ANA. Elevated ESR is seen in most patients, as is hypergammaglobulinemia (180).
Treatment
Ocular and oral disease is treated symptomatically with preservative-free artificial tears, lubricating ointments, methylcellulose drops, good dental hygiene and anti-fungal medications if needed. Muscarinic stimulants such as pilocarpine and cevimeline can aid with salivary gland secretion though side effects such as diaphoresis, nausea, and diarrhea can be limiting. For Sjögren’s associated arthritis, hydroxychloroquine can be helpful. Corticosteroids and immunosuppressives such as cyclophosphamide and azathioprine can be used for severe extraglandular disease, but are not helpful for alleviating symptoms of xerosis (189, 190, 191, 192).
VII. Behçet’s disease
Behçet’s disease is a systemic vasculitis, with an increased prevalence along the old “silk route”, specifically Turkey, Iran and Japan. Though there is no diagnostic test for Behçet’s disease, criteria have been developed by the International Study Group for Behçet’s disease, which include recurrent oral aphthous ulcers (Fig 11.20) as the major criterion, and any two of the following minor criteria: recurrent genital ulcers (most commonly of the scrotum in males (Fig 11.21) and labia majora of women), eye lesions (uveitis, retinal vasculitis), cutaneous findings such as such as erythema nodosum, papulopustules, pseudofolliculitis, acneiform lesions, or a positive pathergy test. The pathergy reaction is characterized by papule or pustule formation one to two days after skin trauma, such as a needle prick. Behçet’s differs from other autoimmune diseases in its lack of B-cell hyperreactivity or T-cell dysfunction, and lack of association with Raynaud’s phenomenon or secondary Sjögren’s syndrome. (193, 194).
Emergent complications
Ocular disease occurs in 70% of cases of Behçet’s disease; approximately 25% may lose their vision despite therapeutic intervention. Males are more likely than females to have severe ocular involvement. Ocular inflammation is the initial manifestation in about 20%. Anterior uveitis is seen more commonly in females, while posterior uveitis is more common in males (195, 196).
An important cause of morbidity in patients with Behçet’s disease is major vessel disease, particularly arterial aneurysm and/or occlusions, hepatic vein occlusion (Budd-Chiari syndrome), and thrombophlebitis of superficial and deep veins of the legs. Behcet’s syndrome is the only vasculitis that causes pulmonary artery aneurysms, which is the primary cause of mortality.
Significant neurological involvement can be seen, including central nervous system disease, especially involving the brainstem, aseptic meningitis, and cerebral venous sinus thrombosis. (197, 198, 199, 200).
Laboratory findings
Laboratory findings are usually nonspecific, and also found in other inflammatory diseases, such as anemia of chronic disease, and elevated acute phase reactants. Autoantibodies are typically negative (194).
Treatment
Mild to moderate mucocutaneous disease can be treated with topical or intralesional corticosteroids, topical sucralfate, and local anesthetics. For more severe cases, colchicine, thalidomide or azathioprine can be used. The nonerosive arthritis of Behçet’s can be managed with colchicine. Ocular disease is treated with prednisone and azathioprine (201, 202). As it is believed that vascular inflammation is the cause of venous thromboses, treatment strategy is aimed toward immunosuppression rather than anticoagulation. Vascular involvement is treated with systemic corticosteroids alone, or in combination with other immunosuppressives such as azathioprine, chlorambucil, cyclophosphamide, cyclosporine, and interferon-. Mycophenolate mofetil, IV immunoglobulin, and anti-TNF- agents have also been used (203, 204).
VIII. Reactive arthritis
Reactive arthritis (ReA), formerly known as Reiter’s syndrome, is a multisystem disease characterized by a sterile oligo- or monoarthritis and either urethritis, cervicitis, or bilateral conjunctivitis following a urogenital or enteric infection. Triggering organisms include Chlamydia trachomatis (or less frequently, Chlamydia pneumoniae), Salmonella, Shigella, Campylobacter, and Yersinia. The pathogenesis is not clearly understood, but it is thought that bacterial components may trigger the disease in genetically susceptible individuals, perhaps via molecular mimicry (205, 206).
There are many classic skin findings. Keratoderma blennorrhagicum (10%) is a scaly, often pustular, eruption that manifests 1-2 months after the start of arthritis, usually involving the soles of the feet (Fig 11.22), but can affect the legs, hands, nails, and scalp. Circinate balanitis (25%) is most frequently seen in Chlamydia associated ReA, and presents as either hyperkerototic plaques on circumcised males, or shallow ulcers on uncircumcised males, typically on the glans penis, or on the shaft or scrotum (Fig 11.23). Yersinia infection can be associated with erythema nodosum. Oral ulcers can also be seen in ReA (205, 207).
Emergent complications
Conjunctivitis is the most common eye finding, but anterior uveitis (iritis), often unilateral, is also seen. Topical glucocorticoid eye drops and a mydriatic agent may be needed to dilate the involved eye and prevent synechiae formation between the pupil and lens.
Acute reactive arthritis may be severe enough to mimic septic arthritis and be associated with high fevers and extreme malaise. Septic arthritis can occur in Campylobacter and Salmonella infections. Furthermore, gonococcal arthritis (a septic arthritis) must be distinguished from an aseptic arthritis arising in a patient developing ReA with a gonococcal infection. Synovial fluid gram stain and culture helps distinguish the two (207).
Laboratory findings
Diagnosis relies on finding the triggering infection. Chlamydia trachomatis can be found in morning urine or by genital swab. By the time arthritis presents, enteric organisms may not be identifiable in the stool. There is a strong association between the genetic marker HLA-B27 and reactive arthritis. Furthermore, HLA-B27 can be predictive of future development of spondyloarthritis and uveitis. (208)
Treatment
Patients infected with C. trachomatis and their sexual partners should be treated with azithromycin or doxycycline.. The arthritis itself can be treated with NSAIDs, and with local glucocorticoid injections for mono- and oligoarthritis and enthesitis. Systemic corticosteroids can be used if there are systemic symptoms. The role of antibiotics in the treatment of reactive arthritis is questionable. Although it may be helpful in the infective phase, once the arthritis has developed, antibiotics typically do not modify the disease course. As half of affected patients recover from reactive arthritis within the first six months, the use of disease-modifying antirheumatic drugs are often not considered, though sulfasalazine may be helpful in achieving faster remission if used in the first few months and for those with chronic reactive arthritis. Cutaneous lesions can be treated with topical steroids (207, 209, 210, 211).
IX. Adult-onset Still’s disease
Adult-onset Still’s disease (AOSD) is an uncommon febrile autoimmune disease, typified by high spiking fevers, polyarthritis, and a transient rash. The etiology of AOSD is unknown, and it typically affects adults in the 2nd and 4th decades of life with equal incidence between males and females. A similar syndrome occurs in the systemic subset of juvenile idiopathic arthritis. Other AOSD symptoms include pharyngitis which can be a presenting symptom, lymphadenopathy, hepatosplenomegaly, leukocytosis, thrombocytosis, elevated transaminases, and serositis (212).
Fever of unknown origin is the most frequent cause for investigation, as high fever is the initial symptom in most patients. The classic fever pattern is one-two spikes exceeding 39° C, most often late in the day, and resolving within hours, also known as quotidian fever (212, 213). AOSD can be mistaken for sepsis, and other disorders such as hepatitis and reactive hemophagocytic syndrome must be excluded. The classic AOSD rash is maculopapular and salmon-colored, mainly over the trunk and proximal extremities, most pronounced during febrile episodes. Dermatographism is present in up to 60% of patients with the rash (212).
Emergent complications
Chronic AOSD can lead to joint destruction akin to seropositive rheumatoid arthritis, with resultant severe functional impairment (212). Up to 5% of patients can develop amyloidosis within 10 years of the disease onset (214). Macrophage activation syndrome is reported in 5-10% of patients. This massive, potentially deadly immune response is associated with cytopenias, disseminated intravascular coagulation, and liver dysfunction (215).
Laboratory findings
AOSD is typically seronegative for ANA and RF. However, acute phase reactants such as ferritin, ESR, and platelets are often elevated. Ferritin levels more than five times the upper limit of normal is specific for AOSD. Serum albumin is commonly low. A majority of patients have leukocytosis, mild anemia of chronic disease and thrombocytosis. Patients with hepatomegaly may have elevated LFTs (212).
Histology of skin lesions is nonspecific with a mild perivascular infiltrate in the superficial dermis consisting of lymphocytes and histiocytes. Myalgias and joint pains also worsen with the rash and fever (212).
Treatment
NSAIDs are first-line treatment for the symptoms of adult-onset Still’s disease, but most affected individuals need oral corticosteroids to treat the acute systemic symptoms. About 20% of patients have chronic relapsing disease, which may require steroid-sparing agents, namely methotrexate for polyarthritis. Cyclosporine, IVIG, biologics such as anti-TNF alpha agents, rituximab, and the IL-1 receptor antagonist anakinra have been used successfully as well (216).
Conclusion
The connective tissue diseases span a broad clinical spectrum with varied presentations in multiple organ systems. As such, the diagnosis of connective tissue disease is difficult, and can be delayed, leading to deleterious end organ effects. The clinical picture is further muddled by the fact that patients with underlying collagen vascular diseases may develop unrelated disease processes, which can mimic a disease ‘flare’ or exacerbation. Cutaneous findings often provide an invaluable clue to diagnosis and management, and are thus critical to preventing the multitude of associated secondary complications.