 Graft-versus-host disease (GVHD) is a multisystem disorder combining features of both autoimmunity and immunodeficiency. It is a common and serious complication of hematopoietic stem cell transplantation (HSCT), and rarely solid organ transplantation, transfusion, and donor lymphocyte infusion. Because of its complex pathophysiology and diverse clinical manifestations, management of this challenging disease requires a multidisciplinary approach. Skin findings are the most common presenting sign, and they found in all forms of GVHD; therefore, the dermatologist plays an important role in diagnosis and therapy.
Graft-versus-host disease (GVHD) is a multisystem disorder combining features of both autoimmunity and immunodeficiency. It is a common and serious complication of hematopoietic stem cell transplantation (HSCT), and rarely solid organ transplantation, transfusion, and donor lymphocyte infusion. Because of its complex pathophysiology and diverse clinical manifestations, management of this challenging disease requires a multidisciplinary approach. Skin findings are the most common presenting sign, and they found in all forms of GVHD; therefore, the dermatologist plays an important role in diagnosis and therapy.
Research into HSCT began in the years following World War II in an effort to address fears over the deleterious effects of radiation exposure.1 Animal research demonstrated that HSCT was able to replace radiation-damaged host cells with healthy, transplanted donor cells. This preclinical data provided impetus for future studies investigating HSCT following chemoradiotherapy in leukemic patients. The use of transplanted cells as a treatment option has driven research into stem cell graft types (autologous, syngeneic, allogeneic) and graft sources (bone marrow, peripheral blood, umbilical cord blood). Improvements in treatment have led to increased use of HSCT for a wide variety of disease. Unfortunately, despite these improvements and research advances, GVHD remains a serious complication of this procedure.
OVERVIEW OF HSCT
The process of bone marrow ablation is termed “conditioning”. Conditioning reduces tumor burden, creating a physical space into which transplanted cells can migrate and proliferate, and it induces a state of immunosuppression so donor stem cells will not be rejected. Myeloablative conditioning consists of total body irradiation and/or chemotherapy. Specific conditioning regimens depend upon center protocols and the disease being treated. Myeloablative conditioning is associated with significant organ toxicity and is usually used in patients under 60 with good performance status. Non-myeloablative, minimally toxic conditioning regimens have been developed for use in older patients or those with significant comorbidities.2 Conditioning usually takes 7 to 10 days.
After completion of conditioning, stem cells are infused intravenously, usually through a central venous line. Stem cells can be derived from bone marrow, peripheral blood or umbilical cord blood. If the patient himself provides stem cells, the transplant is said to be autologous. An allogeneic (allos means other in Greek) transplant means that stem cells are derived from another individual. If the other individual is an identical twin, the transplant is “syngeneic”.
Extraction of bone marrow is usually done under general anesthesia as significant amounts of marrow (>70ml) are obtained from the iliac crests. Extraction of stem cells from peripheral blood is logistically easier, and it avoids the risks of general anesthesia. Peripheral blood is the most common source of allogeneic stem cells in adult recipients (>20 years old), and bone marrow the most common in recipients under 20. Overall, peripheral blood is the most common source of stem cells.3 The donor is primed with granulocyte-macrophage stimulating factor (GMSF), peripheral blood is removed, white blood cells (WBCs) are separated, and red blood cells (RBCs) are reinfused back into the donor. Stem cells can be frozen for prolonged periods without damaging their function or viability. Peripheral blood contains more T cells than bone marrow, thus increasing the risk of graft-versus-host disease (GVHD). 4, 5With umbilical cord blood, there is a lower incidence of GVHD, and a much lower risk of infection by latent viruses such as Epstein Barr (EB) or cytomegalovirus (CMV). One or two HLA disparities can be tolerated by the recipient, and cord blood is easily banked, typed and stored. The main disadvantage is that it contains low numbers of stem cells, and there may be failure of engraftment. Cord blood is mainly used in children.
Following infusion, stem cells migrate to the bone marrow. In the range of 2-4 weeks, they will “engraft” and start producing normal blood cells. The day of engraftment is defined as the first of three consecutive days in which the neutrophil count is greater than 0.5X10(9)/L. If engraftment does not occur by day 30, the graft is considered failed.9 Hematopoietic function normalizes in a matter of weeks; however, immune function takes months, or even years to normalize.10 From the start of conditioning to the time of engraftment, the patient is in a state of pancytopenia and profound immunosuppression. The risk of lethal viral, bacterial or fungal infection is high. Infection can come about from external sources, from endogenous flora of the skin and gut, or through reactivation of latent viruses, particularly CMV.11 Prophylactic antibiotics and careful supportive care are critical during this period. Mucositis can be severe during the pancytopenic phase, and total parenteral nutrition (TPN) may be necessary.12-14 Following engraftment, there is recovery from pancytopenia and healing of mucositis; however, at this early stage the immune system remains weakened and dysregulated.
HSCT is a debilitating, emotionally taxing, high-risk procedure associated with significant treatment-related morbidity and mortality.15, 16 In the months to years it takes for the engrafted stem cells to differentiate, proliferate and give rise to a functional immune system, patients suffer infections, fatigue and psychological debility. Children and young adults are most likely to experience complete immune reconstitution; however, complete immune recovery may never take place in older adults, particularly in patients who develop chronic GVHD (cGVHD). 17, 18 Following HSCT, donor-derived, immunologically-active cells are confronted with infection, residual malignant cells, and a variety of foreign proteins. Acute GVHD (aGVHD) results from this confrontation. Acute GVHD is a major cause of morbidity and mortality following HSCT.19
Acute GVHD occurs when donor T cells react against recipient HLA antigens and minor histocompatibility antigens (mHA). It occurs in 60-80% of patients in which there is an HLA mismatch, and in 20-50% of patients who receive stem cells from an HLA identical sibling donor.20 In 1966, Rupert Billingham identified three preconditions that need to be met in order for GVHD to occur.21 One, the graft must contain immunologically active cells, now known to be alloreactive T cells. Two, the recipient must express tissue antigens foreign to the donor. Three, the recipient must be incapable of mounting an immunological response against the donor cells. The incidence and severity of aGVHD relate to the extent of mismatch of HLA proteins.19 Ideally, there will be high resolution matching for HLA-A, B, C and DR B1 between donor and recipient (“8/8 match”).19 However, even if the donor is an identical twin, GVHD can still occur because of mismatch of minor histocompatibility antigens (mHA).22, 23
Every human being is genetically unique, and this uniqueness also applies to monozygotic twins in whom subtle, but medically significant genetic differences exist. For example, single nucleotide polymorphisms (SNPs) in the thiopurine methyltransferase gene have been identified in identical twins. These polymorphisms result in differences in metabolizing capacity between the twins. These genetic differences come about because of somatic mutations that occur in utero, meiotic and mitotic recombination and biased gene conversions. Genes and genomes are in a state of constant evolution. 22, 24
Minor HAs are immunogenic peptides derived from genes outside the MHC. They arise through polymorphisms in homologous genes between recipient and donor.25 Polymorphisms in recipient genes result in different amino acid sequences, which are recognized by donor T cells as non-self.19 Approximately 30 mHAs have thus far been identified in humans.22 For example, males express several H-Y antigens encoded on the Y chromosome. These antigens are recognized as foreign by lymphocytes from female donors, and aGVHD will result from gender mismatch.26 Multiple other mHAs exist, all of which can trigger aGVHD.27 Minor HAs may play an important role in graft-versus-leukemia (GVL) effect because some of these antigens are only expressed on hematopoietic cells.28 The success of HSCT in leukemia is due in part to GVL effect mediated by donor immune cells.25, 28 The relapse rate of leukemia is lower in patients who develop GVHD compared to those who do not. It is also lower in patients receiving allogeneic versus syngeneic transplants because the former are more likely to develop GVHD than the latter.
ACUTE GRAFT VERSUS HOST DISEASE
GVHD has traditionally been divided into two forms, acute and chronic. Acute GVHD classically referred to disease that occurred in the first 100 days, and chronic, after day 100. This classification has proven inadequate because aGVHD can occur after day 100, particularly in patients who have undergone reduced intensity conditioning, and features of cGVHD are common in the first 100 days. This temporal division has been superseded by a new classification developed by experts in an NIH Consensus Development Project on Criteria for clinical trials in chronic graft-versus-host disease.29 In the new classification, the two main categories remain, but each has two subcategories. Acute GVHD is subclassified into “classic” aGVHD occurring before day 100 following the transplant, or persistent, recurrent or late aGVHD occurring after day 100. Chronic GVHD is subclassified into classic cGVHD, and an overlap syndrome in which features of acute and chronic GVHD occur together. In the new classification system, clinical and pathologic manifestations take priority over time of onset.
Ferrara and associates have identified three distinct phases in the pathogenesis of aGVHD. Phase one involves induction of host cytokines and activation of host antigen presenting cells (APCs). Infections, conditioning regimens and underlying disease all cause cellular damage and release of pro-inflammatory cytokines such as TNF-α, IL-1, and IL-6. Increased serum levels of these cytokines upregulate adhesion molecules, MHC molecules and mHAs on APCs.30 In addition, damage to the GI tract from conditioning results in translocation of immunostimulatory molecules such as lipopolysaccharide (LPS) and other bacterial products into the systemic circulation.32, 33 Macrophages and other cells of the innate immune system recognize pathogen associated molecular patterns, and they secrete cytokines, chemokines, adhesion molecules and costimulatory molecules. The innate immune system is “primed”. MHC molecules are upregulated, leading to enhanced antigen presentation by antigen-presenting cells.34 Decontamination of the gastrointestinal tract has been associated with reduced incidence of aGVHD both clinically and in animal models, and in some centers, gut decontamination is standard procedure.34
Phase two consists of antigen presentation to donor T cells. This occurs in recipient lymphoid tissue. In direct presentation, donor T cells recognize peptides bound to allogeneic MHC molecules on recipient APCs, or they recognize and react to the allogeneic MHC molecule itself without peptide.35 In indirect presentation, donor APCs will degrade allogeneic MHC molecules into peptides, which are presented on the cell surface by donor-MHC molecules and recognized by donor T cells. In response to antigen presentation, activated donor T cells proliferate and differentiate. They then exit lymphoid tissue and traffic to target organs. Activated T cells secrete cytokines, in particular IL-2 and IFN-γ, both of which are important mediators of aGVHD. These cytokines trigger cytotoxic T lymphocyte (CTL) responses to alloantigens, and they also induce monocytes and macrophages to produce proinflammatory cytokines IL-1 and TNF-α.35
During phase three, the effector stage, actual target organ damage takes place.30, 35 CTLs are the predominant cellular effectors of acute GVHD, and they cause tissue damage via the Fas/FasL and the perforin/granzyme pathway. TNF-α, IL-1 and nitric oxide (NO) are the main inflammatory cytokine effectors. TNF-α activates APCs, it enhances alloantigen recognition, and it recruits effecter cells to target organs.19 It also directly causes tissue damage by inducing apoptosis of target cells after activation of the TNF-Fas activation pathway.36 NO, produced by macrophages, inhibits proliferation of epithelial stem cells and thereby inhibits repair of target tissue.35 Damage to the GI tract occurring during phase three causes more release of bacterial LPS, which further upregulates innate immune function. A feedback loop is created that enhances antigen presentation and activation of alloreactive donor T cells.30
Natural killer (NK) cells appear to modulate aGVHD. NK cells are the first lymphocytes to recover to normal numbers after HSCT, and they play a role in both innate and adaptive immunity. Early animal models demonstrated that NK cells were not crucial for the development of GVHD and their role in GVHD was incompletely understood.37 More recently, NK cells have been shown to decrease GVHD severity by inhibiting immature dendritic cells and suppressing alloantigen-driven proliferation of donor CD4+ T cells.38, 39 In addition, they can work in concert with T regulatory (Treg) cells to prevent chronic GVHD.39 NK cell function may be more important in unrelated donor HSCTs compared to related donors as polymorphisms in NK cell receptor and ligand complexes have been linked to differences in GVHD and relapse rates. Patients undergoing unrelated HSCT with a graft containing high numbers of NK cells (CD56+) had a significantly reduced incidence of severe aGVHD compared to patients receiving grafts with lower numbers of NK cells.40 This effect was not seen with related donors and did not seem to influence the graft-versus-leukemia (GVL) effect.
Acute GVHD (aGVHD) can involve many organs; however, skin, intestine and liver are the main targets.20 Skin is usually the first organ involved with rashes occurring in at least 80% of patients.20 Hepatic and intestinal manifestations appear a few days after the rash.41 Extensive rashes are common in the post-transplant period, and these can include drug reactions, chemotherapy related toxicity, viral exanthems, toxin-mediated erythemas, engraftment syndrome, and intrinsic inflammatory skin diseases not related to the HSCT. These pathologic processes may occur simultaneously.
Dermatologists play a key role in sorting out post-transplant rashes. Diagnosis of aGVHD is based on clinical and histopathologic features. The main histopathologic features of cutaneous aGVHD include a lymphocytic interface dermatitis, vacuolar degeneration of the basal cell layer, and necrotic keratinocytes.42 Histopathologic grading is based on the extent of keratinocyte necrosis, which ranges from subtle vacuolar alteration to complete epidermal necrosis. A skin biopsy can yield valuable diagnostic information with minimal risk to the patient. In contrast, invasive endoscopic procedures and biopsies of intestine and liver carry significant procedural risks, particularly with respect to bleeding and infection post-transplant., 23, 43,44
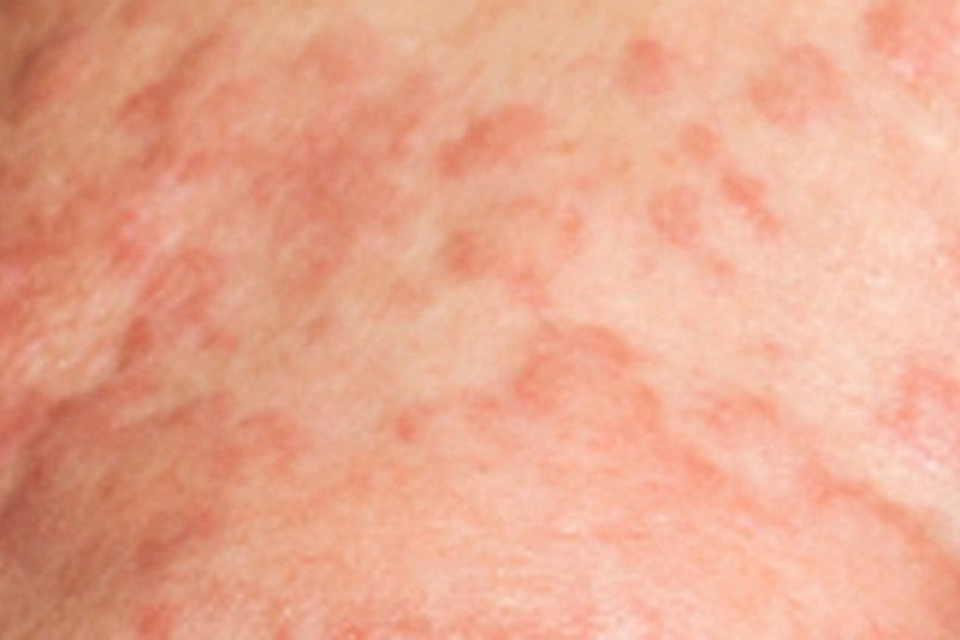
Acute GVHD usually manifests two to six weeks after transplantation. As a rough rule of thumb, rashes occurring in the first 20 days post-transplant are more likely drug reactions, reactions to conditioning, or infections rather than aGVHD. Acute cutaneous GVHD is heralded by a pruritic, sometimes painful “maculopapular” rash that tends to be acrally distributed. (Fig 6.4, 6.5, 6.6) Involvement of the palms, soles, pinnae and cheeks is common.45 Involvement of the face, palms and soles together is strongly predictive of aGVHD rather than drug reaction.46 The rash of aGVHD commonly affects the upper, sun-exposed areas of the body such as the face, arms and shoulders.43, 47 Exanthematous drug reactions usually begin centrally and often do not involve the palms and soles. A follicular localization is also typical of aGVHD and an important clue to diagnosis.20, 48-50 Atypical and severe skin manifestations of aGVHD may occur.51 Acute grade IV cutaneous GVHD resembles SJS/TEN, and mucositis consisting of oral and lip erosions occurs in approximately 60% of these patients.51 However, in milder forms of aGVHD, mucositis is not a prominent clinical feature.47 Other mucosal manifestations of acute GVHD include erythema and lichenoid lesions.
The liver is a common target organ in aGVHD. Abnormal liver functions and jaundice secondary to cholestasis comprise the typical clinical picture. Biliary epithelial cells are the primary target of alloreactive donor T cells recognizing foreign HLA antigens and minor HAs on the surface of these cells.52 Histologically, there is lymphocytic infiltration of small bile ducts, nuclear pleomorphism, and apoptosis of epithelial cells.53 Untreated, acute hepatic GVHD can lead to loss of bile ducts and worsening jaundice. A rising or persistent elevation of the bilirubin portends a poor prognosis. In some patients aGVHD can present with a hepatitis-like picture with elevation of aminotransferase enzymes.52, 53 Fifty percent of patients with acute hepatic GVHD will develop chronic hepatic GVHD, and only 30% of patients with acute hepatic GVHD will experience complete resolution of liver abnormalities after initial immunosuppressive treatment.52 The differential diagnosis of liver abnormalities in the post-transplant patient is complex and includes sinusoidal obstruction syndrome (SOS), acute viral hepatitis and other infections, biliary obstruction, drug induced liver injury, TPN hepatotoxicity, and numerous other conditions.53, 54 Acute hepatic GVHD usually does not occur in isolation, and other stigmata of aGVHD such as rash and GI abnormalities are part of the total clinical picture.
Timing is also an important consideration. If signs of liver damage occur in the first 20 days post-transplant, causes other than aGVHD are likely, especially SOS. SOS occurs following damage to the hepatic sinusoids from myeloablative conditioning, especially regimens containing cyclophosphamide, the metabolites of which are toxic to sinusoidal endothelial cells.52 The condition is characterized by varying degrees of hepatomegaly, fluid retention, jaundice and biochemical abnormality. Severe cases can be fatal.
The predominant clinical sign of acute intestinal GVHD is diarrhea. Vomiting, anorexia, abdominal pain and ileus may also be present. Diarrhea in the first 20 days after HSCT is usually associated with conditioning toxicity. From day 20 to day 100, aGVHD is most likely, and after day 100, an enteric infection should be considered.44 Biopsy of intestinal mucosa in acute intestinal GVHD will show inflammatory changes associated with epithelial cell apoptosis, the presence of which is necessary for histopathologic diagnosis.44 The rectosigmoid area is the optimum site for biopsy. In comparison to other sites in the GI tract such as stomach or duodenum, rectosigmoid mucosal biopsies have a sensitivity of 95.6% and a specificity of 100%.55
In the post transplant period, a disorder known as engraftment syndrome (ES) can occur. ES shares some clinical features with classic aGVHD, and some authorities consider ES a variant of aGVHD. ES occurs after allogeneic or autologous HSCT at the time of neutrophil recovery.56 Major features consist of generalized maculopapular rash, fever, and noncardiogenic pulmonary edema associated with hypoxemia. The clinical picture can be accompanied by diarrhea, weight gain secondary to fluid retention, hepatic or renal dysfunction, transient encephalopathy and keratitis.57, 58 The reported incidence of ES varies from 7 to 59%. The wide disparity in numbers probably reflects differences in definition. In one large series of 328 patients undergoing peripheral blood autologous HSCT, the incidence was 12.8%.57
ES is thought to result from release of proinflammatory cytokines and products of degranulation and oxidative metabolism of neutrophils.59 This results in systemic endothelial cell damage.57, 59 ES responds well to corticosteroids when given in a timely fashion and in adequate amounts; however, if treatment is delayed or inadequate, ES can progress to multiorgan failure and death.60 Patients with ES have a 21% greater likelihood of non-relapse mortality in five years compared to those without ES. Steroid treatment does not reduce this increased mortality.60
Autologous GVHD (autoGVHD) was first reported by Hood et al in 1987.61 Seven patients receiving autologous bone marrow transplantations developed cutaneous eruptions compatible both clinically and histologically with grade II aGVHD. Also known as the “auto-aggression” syndrome, autoGVHD tends to be milder than aGVHD following allogeneic HSCT. It usually involves skin alone and is generally self-limited; however, more severe forms involving the liver and intestine can occur. AutoGVHD is thought to result from loss of self-tolerance because of loss of regulatory T cells (Treg) and damage to the thymus from conditioning.62 Treg cells are a subset of T cells that prevent self-reactive T cells that have escaped thymic deletion from reacting to self antigens. Treg cells establish tolerance to both allo- and self MHC antigens.63 Myeloablative conditioning causes depletion of Treg cells.64 In addition, cyclosporine (CSA) and other medications administered post-transplant interfere with the functional capacity of the thymus to delete T cells bearing autoreactive TCRs. This effect has been demonstrated in rodents, which are considered accurate models of human aGVHD.64, 65 In the setting of autologous HSCT, loss of central tolerance coupled with the absence of peripheral Treg cells creates a permissive environment in which autoreactive T cells damage host tissue.62
Although aGVHD causes considerable morbidity and mortality, it is associated with significant anti-tumor effects. It has been observed repeatedly that patients with aGVHD have lower disease relapse rates than those without. Because autoGVHD tends to be mild and limited to skin, investigations have been undertaken to harness graft-versus-tumor (GVT) effect by artificially inducing autoGVHD by post-transplant administration of CSA alone, or CSA plus IFNγ or IL-2. Unfortunately, no clear-cut anti-cancer benefit has thus far been shown in clinical trials.64, 66
ACUTE GVHD FOLLOWING SOLID ORGAN TRANSPLANTATION, TRANSFUSION AND DONOR LYMPHOCYTE INFUSION
Acute GVHD after solid organ transplantation is an uncommon, but serious complication with high mortality. It can occur with transplantation of any solid organ, but it is most likely to occur after small intestine transplant (5-10% incidence) or orthotopic liver transplant (OLT) (1-2% incidence).67 (“Orthotopic” means the donor organ is transplanted into the same place as the patient’s original organ.) OLTs are performed much more commonly than intestinal transplants, so aGVHD associated with solid organ transplants generally refers to OLT.68 Alloreactive lymphocytes are transplanted along with the organ, and liver and intestine contain the largest numbers. In fact, the number of lymphocytes transferred in an OLT is roughly equivalent to that in a bone marrow transplant.69
Despite the large number of passenger lymphocytes, the incidence of GVHD is low. An important reason is that a high percentage of hepatic T cells express γδ receptors, 7-54% versus
<6% in peripheral blood. These cells produce Th2 cytokines, which downregulate Th1 responses and suppress aGVHD.70 In addition, patients receiving OLTs are immunosuppressed, not immunoablated. Donor T cells are rejected by a partially functioning immune system.
Skin rash is the major clinical sign associated with OLT-associated aGVHD.71 Lesions erupt one to eight weeks after transplantation. Skin lesions usually begin distally on the palms and soles and rapidly generalize.70 Rapid progression to the GI tract and bone marrow follow.70 Fever in association with the rash is a poor prognostic sign.
Acute GVHD associated with OLT differs from aGVHD following HSCT in several respects. The transplanted liver itself is not a target organ because alloreactive lymphocytes recognize transplanted liver tissue as self. On the other hand, bone marrow involvement manifesting as pancytopenia is a common and serious development. 70 Neutropenia can be a presenting sign of GVHD following OLT.70, 71 Because of high mortality, and immunologic rejection of donor T cells, chronic GVHD following OLT is unusual. 70
A risk factor for aGVHD in the setting of OLT is HLA compatibility.68 The more well-matched the graft, the greater the likelihood of aGVHD. If alloreactive hepatic T lymphocytes express shared HLA determinants with the recipient, they are less likely to be destroyed by the recipient immune system.70 Age greater than 65, and large disparity in age between a young donor and an older recipient are also risk factors for the development of aGVHD following OLT.
Given the rarity of aGVHD following solid organ transplantation, diagnosis can be challenging. The usual presenting sign is rash, sometimes associated with fever. The differential diagnosis includes viral exanthem or other infection, drug eruption, and aGVHD.72 A skin biopsy showing interface dermatitis, vacuolar degeneration of basal cells and necrotic epidermal cells is highly suggestive of GVHD. 70 Donor cell chimerism is also an important feature of GVHD associated with solid organ transplantation. Chimerism refers to the presence of a mixed population of donor and recipient lymphocytes in tissue or peripheral blood. Although important, chimerism is not absolutely diagnostic. It is not present in all patients with solid organ aGVHD, and it is a frequent finding immediately after OLT.73 Clinical response to treatment correlates with decrease or disappearance chimerism.69
Transfusion associated aGVHD (TA-GVHD) occurs when transfused donor lymphocytes engraft in an immunocompromised host, or when the donor is homozygous for an HLA class 1 haplotype of the recipient, which allows donor lymphocytes to escape immunologic recognition by the recipient.74 This sometimes happens when blood relatives donate blood to immunocompetent recipients.74-76 Certain clinical situations are also high risk. Granulocyte transfusions contain large numbers of lymphocytes, and patients receiving them are usually neutropenic and immunocompromised. Infants receiving exchange transfusions for erythroblastosis fetalis receive a large volume of blood and cells, and their immune systems are immature. Patients with Hodgkin’s disease, non-Hodgkin lymphoma (NHL), congenital immunodeficiency diseases, persons undergoing HSCT, and persons treated with purine analogues such as cytarabine are at risk for TA-aGVHD.74, 75, 77 Patients with solid malignancies, hematologic malignancy other than Hodgkin’s disease/NHL, and patients with AIDS are not considered to be at high risk.74
TA-aGVHD begins abruptly with fever 3 to 30 days after the transfusion. Fever is followed by a generalized maculopapular rash, which can progress to erythroderma and bulla formation. Profuse diarrhea, vomiting, anorexia, right upper quadrant pain, jaundice, elevated liver enzymes, and pancytopenia complete the clinical picture.75 The overall tempo of TA-aGVHD is more rapid than in HSCT-associated aGVHD. 75 Diagnosis is made by clinical signs and symptoms, the demonstration of chimerism in blood and tissue, and skin biopsy findings of interface dermatitis, basal cell vacuolization, and epidermal cell necrosis.
Unfortunately, treatment for TA-GVHD is generally ineffective, and the overall mortality exceeds 90%.74 Prevention is the best and most effective management strategy. Prevention consists of identifying those at risk. Once identified, these patients should only receive blood and blood components that have been irradiated with 2000cGy or higher.74, 75 Irradiation of blood and blood products blocks alloreactive lymphocyte proliferation, but it does not interfere with the function of RBCs, WBCs or platelets. Although some leakage of potassium has been observed in irradiated blood, there is no clinical evidence that irradiated blood is harmful to patients.74, 78
Should leukemia relapse after HSCT, infusion of lymphocytes from the original donor can be used as a salvage treatment. Donor lymphocyte infusion (DLI) relies on graft-versus-leukemia effect, and it has proven quite effective in certain forms of leukemia, particularly in relapsed chronic myelogenous leukemia (CML).79 Complete molecular and cytogenetic remissions have been achieved using this modality in a substantial proportion of treated patients.80 Acute and chronic GVHD can complicate DLI; however, GVHD following DLI tends to be mild to moderate, it is more responsive to treatment with immunosuppressive therapy, and the onset tends to occur later than GVHD following HSCT.79 The reason is that the tissue damage and “cytokine storm” resulting from conditioning, infection and underlying disease are generally not present in the setting of DLI. However, the presence of either acute or chronic GVHD following DLI is associated with a 2.3 fold risk of death in comparison to patients without GVHD.81 Chronic GVHD develops in 33% to 61% of patients following DLI.82
STAGING AND GRADING OF ACUTE GVHD
Stratification of aGVHD severity is important in determining prognosis. It is also useful in determining entry into clinical trials and assessment of outcomes to particular regimens. In 1974, Glucksberg proposed a grading system for aGVHD severity.83 The Glucksberg system is intuitive, easy to apply, and has been in common use ever since. Severity of skin, liver and intestinal tract involvement is measured and assigned stages from one to four. To determine grade, the stages of the three organ systems, and a subjective one to four assessment of overall performance status are tabulated. The patient is then assigned a grade of severity from one to four. In the Glucksberg system there are 125 possible combinations when three organs plus performance status are staged one to four.
The International Bone Marrow Transplant Registry (IBMTR) severity index was created after it was noted that patients with the same Glucksberg grade but with different patterns of organ involvement had different outcomes.85 For example, patients with grade II Glucksberg, but stage 3 skin involvement had significantly higher risk of death than patients with grade II Glucksberg with stage 0-2 skin involvement.85 The IBMTR severity index assigns letter grades from A to D. The assigned grade is based on the maximum involvement in an individual organ regardless of involvement of other organ systems.85 It relies only on objective measurement of organ involvement, thereby reducing inter-observer variability. Thus, in the example above, a patient with stage three skin involvement would be assigned to level C even if liver and intestinal tract were stage one. In addition, the IBMTR severity index eliminates subjective assessment of performance status.
In a mixed retrospective and prospective study of 114 patients, Martino et al found the IBMTR index superior to the Glucksberg grading system in predicting transplant related mortality and failure.86 However, in a prospective study of 607 patients evaluating the two systems, Cahn et al observed strong agreement between the two systems and no clear-cut advantage of one over the other.84
PROPHYLAXIS AND TREATMENT OF ACUTE GVHD
Risk factors for the development of aGVHD include histoincompatibility of MHC antigens (particularly class II antigens), older age of donor and recipient, gender mismatch (particularly female donor with male recipient), presence of infection, underlying disease stage and intensity of conditioning.9, 26 To the extent possible, these risk factors should be modified. In addition, use of cord blood and decontamination of the gut are associated with lower incidence of aGVHD.6, 32
GVHD prophylaxis usually consists of a combination of calcineurin inhibitor (CNI) plus methotrexate or mycophenolate mofetil (MMF).23 The purpose of prophylaxis is to inhibit the activity of alloreactive T cells. Without prophylaxis the incidence of aGVHD approaches 100%.27 Tacrolimus appears to be somewhat superior to cyclosporine, and is less toxic.27MMF is associated with less mucositis than methotrexate. Antithymocyte globulin and anti-CD52 monoclonal antibody (alemtuzumab) have also been used for prophylaxis. Ex-vivo depletion of T cells in the donor graft is associated with decreased incidence of aGVHD, however this technique is associated with increased incidence of malignant disease relapse.27
First line treatment of aGVHD consists of corticosteroids.23, 27, 30 Patients are usually started on methylprednisolone 2mg/Kg/day, and the dose is slowly tapered depending on clinical response. Roughly 25% of patients will experience a complete response, and 50% a partial response. For corticosteroid nonresponders, a wide variety of therapies has been tried including PUVA, extracorporeal photophoresis, anti-TNF agents, anti-CD3, anti-IL2 receptor, denileukin difitox, sirolimus, antithymocyte globulin, pentostatin, and visilizumab, an anti CD-3 monoclonal antibody.23, 27, 30, 87 Corticosteroid non-responders have a poor prognosis.
CONCLUSION
HSCT ranks as one of the most radical interventions in the whole of medicine. The patient is given nothing less than an entirely new immune system, and he is potentially cured of hitherto untreatable, fatal diseases. New indications for HSCT are constantly arising, and the procedure is being performed with greater frequency. As with all drastic interventions, HSCT comes with a high cost: aGVHD. At high severity grades, aGVHD is a direct threat to life. Even with the best of care, mortality is high. Lower severity grades are associated with considerable morbidity, and there is a high chance of evolution into cGVHD. Chronic GVHD in its own right is a serious, devastating disease associated with a huge panoply of medical and psychological problems. On the other hand, aGVHD is associated with GVT effect. A consistent observation is that patients with GVHD have a lower rate of leukemia and cancer relapse than those who do not. Thus far, all interventions that ameliorate or prevent GVHD lessen GVT effect. The “holy grail” of treatment is to separate the two by targeting GVHD while preserving GVT. This remains an area of intensive, ongoing research.
CHRONIC GRAFT VERSUS HOST DISEASE
Chronic GVHD (cGVHD) can arise during or directly after an episode of aGVHD (progressive onset cGVHD), after a disease-free interval (quiescent cGVHD), or it can present de novo.88 The condition usually occurs within three years after transplant, with an average onset of five months.89 Chronic GVHD is a multisystem disorder of immune dysregulation combining features of autoimmunity and immunodeficiency. It can affect virtually any organ, and serious infection and secondary malignancy are constant threats. Chronic GVHD profoundly damages quality of life, and patients with the condition suffer a variety of disabilities and impairments. Chronic GVHD is associated with graft versus tumor (GVT) effect, a phenomenon in which alloreactive T cells attack host cancer cells in addition to normal tissue. Because of GVT effect, the incidence of cancer relapse is lower in patients who experience cGVHD. Ideally, treatment regimens for malignant disease aim to preserve GVT effect while preventing GVHD.
As previously stated, a 2005 NIH consensus conference established new guidelines for the diagnosis and staging of GVHD.29 The new guidelines emphasized that clinical manifestations rather than time are the major criteria by which cGVHD is distinguished from late aGVHD. The clinical manifestations of cGVHD are classified into four categories: diagnostic, distinctive, common and other. Diagnostic features are considered pathognomonic for the disease, and when present they establish the diagnosis without any further testing. Diagnostic skin findings include poikiloderma, lichen planus-like eruption, deep sclerotic lesions, morphea-like lesions and lichen sclerosus-like lesions. Distinctive lesions are those commonly seen in cGVHD, but not sufficiently unique to be considered diagnostic. These include depigmentation, sweat impairment, pruritus, maculopapular rash and erythema. Distinctive features require further testing such as skin biopsy, or demonstration of cGVHD in another organ system. Common features are those that occur in both acute and chronic GVHD and include maculopapular rash, pruritus and erythema. Other features such as keratoses pilaris, hyperpigmentation and ichthyosis are nonspecific and cannot be used to establish the diagnosis. The diagnosis of cGVHD requires at least one diagnostic clinical sign, or one distinctive clinical sign plus a positive diagnostic test, and the exclusion of other possible diagnoses, particularly late-onset aGVHD, malignancy, drug reaction and infection. An ‘overlap’ subtype encompasses features of both acute and chronic GVHD.
The reported incidence of cGVHD following allogeneic HSCT varies, but the overall incidence is in the range of 50% to70%.90, 91 Incidence depends on multiple predisposing factors including prior acute GVHD, older recipient age, intensity of conditioning, hematopoietic stem cell source (peripheral blood > bone marrow > umbilical cord blood), mismatched or unrelated donor, female donors with male recipients, graft manipulation (T-cell depletion), GVHD prophylaxis regimen and use of post-transplantation donor lymphocyte infusions.29, 89, 92 In a multi-center cohort analyzing 5,343 HCST recipients with chronic GVHD, the probability of overall survival was 72% at 1 year and 55% at 5 years with a non-relapse mortality (death unrelated to the original cancer) of 21% and 31% at 1 and 5 years, respectively.89 Using multivariate analysis of overall survival and non-relapse mortality, the study further classified patients into risk categories and found several variables associated with a poorer outcome. A prospective study comparing outcomes of patients with ‘overlap’ GVHD versus those with classic cGVHD showed that overlap patients had greater functional impairment, higher symptom burden and increased non-relapse mortality.93
Chronic GVHD most commonly affects skin, mucosal surfaces, eyes, musculoskeletal system, gastrointestinal tract, lungs, liver, and bone marrow. Clinical findings often mimic autoimmune diseases, particularly scleroderma, systemic lupus erythematosus, and Sjögren’s syndrome. Sclerodermoid, lichenoid lesions and lichen-sclerosus-like lesions are the characteristic skin manifestations of advanced cGVHD; however, subtle and nonspecific skin changes such as xerosis, maculopapular rash, and ichthyosis can characterize early disease.
Sclerotic variants range from superficial morphea or lichen-sclerosus-like lesions to deep, diffuse indurated plaques resembling scleroderma. An association exists between exposure to total body irradiation and the development of sclerotic cGVHD.94 All forms of sclerotic GVHD can progress to disfigurement and impairment. Sclerosis of underlying subcutaneous tissues can lead to edema, joint stiffness, decreased range of motion, and contracture. Lichenoid cGVHD presents with violaceous papules and plaques on extensor surfaces. The clinical presentation is virtually identical to that of idiopathic lichen planus. Other skin changes associated with cGVHD include depigmentation, poikiloderma, hypohidrosis with heat intolerance, erythema and pruritus.95 Pigment abnormalities can cause severe cosmetic disfigurement. Chronic cutaneous GVHD can be associated with an isomorphic or isotopic response. Sclerotic cGVHD often appears in areas of minor skin trauma (isomorphic response, or Koebner phenomenon), and lichenoid cGVHD sometimes follows another skin disease such as herpes zoster (isotopic response).95, 96
The main histopathologic features of lichenoid cGVHD lesions consist of hyperkeratosis, focal hypergranulosis, vacuolar degeneration of the basal cell layer and a band-like infiltrate of lymphoid cells. Sclerodermatous lesions show epidermal atrophy and fibrosis. Fibrosis can involve the dermis alone, or it can also involve subcutaneous fat and muscle. Lichen-sclerosis like lesions show epidermal atrophy, hyperkeratosis with follicular plugging and edema coupled with homogenization of the upper dermis. All three chronic cutaneous GVHD variants closely resemble both clinically and histologically their respective dermatologic conditions.
Oral and genital disease occurs in up to 80% patients.97 Lichen planus-like lacy white plaques, ulceration, and scarring occur on the oral and genital mucosa of both women and men; however, genital lesions cause more functional impairment and more severe symptoms in women. Vulvovaginal lesions cause dyspareunia and interference with sexual function. Genital lesions are usually associated with concurrent oral involvement.98 Chronic GVHD commonly affects salivary glands with resultant xerostomia.99,100
Ocular manifestations of cGVHD include xerophthalmia, blepharitis, corneal ulcer, cicatricial conjunctivitis, scleritis, glaucoma and infections. Xerophthalmia is the most common problem. Diagnosis is based on an abnormal Schirmer test and on slit-lamp demonstration of keratoconjunctivitis sicca. Chronic ocular GVHD rarely affects visual acuity, and it is generally accompanied by other systemic findings 29, 101
Nail changes are present in up to 50% of patients, and longitudinal ridging is the most common finding.102 Periungual erythema, brittle nails, Beau’s lines, pterygium, and complete nail loss are also features of cGVHD. Dermatoscopy reveals abnormalities in nailbed capillaries of patients with sclerodermoid cGVHD, but not in patients with lichenoid cGVHD. These abnormalities include enlarged capillaries, neovascularization, avascular areas and hemorrhage.103 Both scarring and nonscarring alopecia occur in cGVHD, and poliosis is common. Hair abnormalities associated with cGVHD should be distinguished from chemotherapy or radiation effects.
Systemic cGVHD often involves the GI tract, liver, lungs and immune system. Nausea, vomiting, diarrhea, anorexia, weight loss, and abdominal pain are typical manifestations of gastrointestinal cGHVD. Endoscopic biopsy and characteristic radiographic findings confirm the diagnosis. Chronic hepatic GVHD is usually associated with cholestasis, elevated bilirubin and elevated alkaline phosphatase. Definitive diagnosis requires positive liver biopsy findings plus a distinctive cGVHD manifestation in another organ system.
Pulmonary signs and symptoms include cough, wheezing, and dyspnea on exertion. These result from bronchiolitis obliterans (BO), an airway disorder in which bronchioles become obstructed by fibrous granulation tissue. BO is a diagnostic pulmonary manifestation of cGVHD, and diagnosis of BO is confirmed by biopsy and abnormal pulmonary function tests.29 Lung dysfunction is serious, and its presence is invariably associated with the highest severity grades of cGVHD.104
Thrombocytopenia and other cytopenias are harbingers of decreased survival.105 Eosinophilia is common in cGVHD. Once thought to be a favorable predictor, reports now show no correlation between eosinophilia and outcome.106 The effect of cGVHD on immunity can be devastating, and infection is a common cause of death. Immunosuppressant and immunomodulating medications worsen susceptibility to infection.
The pathophysiology of chronic GVHD is complex and current knowledge remains incomplete. A primary reason is the lack of animal models that recapitulate human disease as completely in cGVHD as they do in aGVHD.107, 108 Several mechanisms have been identified that alone or in concert contribute to pathogenesis.88 Damage to thymic epithelium from conditioning and aGVHD interfere with thymic deletion of self-reactive T cells, resulting in loss of self-tolerance. Failure of central tolerance can lead to autoimmune phenomena.
Under physiologic conditions, not all self-reactive T cells are eliminated in the thymus, and some make their way to the periphery. A subset of T cells, regulatory T cells (Treg), inhibits self-reactive T cells that have escaped thymic deletion from proliferating in response to self-antigens. Treg cells exert their effects through several cytokines including TGF-β and IL-10. Treg cells express CD4+CD30+ and the transcription factor forkhead box P3 (FoxP3).In cGVHD, the numbers of CD4+CD30+FoxP3+ Treg cells are diminished.109, 110 Strategies to increase numbers of Treg cells such as extracorporeal photophoresis and low dose IL-2 are associated with clinical improvement.91, 111
B cell homeostasis is disturbed in cGVHD, and both host-derived and donor-derived B cells appear to play a role in pathogenesis.109, 112 113 The importance of B cells in cGVHD is evidenced by significant clinical improvement following use of rituximab, an anti-CD20 monoclonal antibody causing B cell depletion. Several case series of patients treated with rituximab for cGVHD have shown improvement with response rates varying from 43% to 80%. Manifestations of skin, mucosal and musculoskeletal cGVHD respond best. 90, 114 B cells perform multiple immunologic functions other than antibody production. B cells present antigen, produce cytokines and chemokines, and regulate immune reactivity.88, 90 Autoreactive B cells in cGVHD secrete proinflammatory cytokines including IL-6, TNF-α, and IFN-γ that activate T cells, macrophages and NK cells.90 The net result is potentiation of the inflammatory cascade. Antigen presentation by autoreactive B cells promotes autoimmune tissue damage independent of antibody production.90 Patients with cGVHD have a higher frequency of autoantibodies, including antinuclear antibodies and antibodies to smooth muscle, cardiolipin, dsDNA, and platelet-derived growth factor receptor (PDGFR).115, 116 It is not entirely clear whether these autoantibodies are directly pathogenic or merely bystanders. Transfer of autoantibodies from cGVHD mice does not produce disease in normal mice.90 However, autoantibodies to PDGFR have a stimulatory effect on fibroblasts and induce production of collagen. Stimulatory antibodies to PDGFR were found in 22 patients with extensive cGVHD, but not in HSCT patients without GVHD or in normal controls. Levels were highest in those with generalized skin involvement and lung fibrosis. Autoantibodies to PDGFR may contribute to fibrosis and the scleroderma-like phenotype in cGVHD.117
B cell-activating factor of the TNF family (BAFF), also known as B-lymphocyte stimulator, is a cytokine that provides survival signals to B cells and prevents apoptosis.90, 113 High levels are present in autoimmune diseases such as SLE, and a monoclonal antibody against BAFF (belimumab) is used in treatment of active lupus.90 High levels of BAFF are also present in cGVHD. Chronic GVHD is associated with low numbers of naïve B cells and high levels of activated, autoantibody-producing CD27+ B cells.118 The high levels of BAFF enhance survival of activated alloreactive and autoreactive B cells, thus causing perpetuation of disease.
Chronic GVHD in humans and in murine models is associated with expansion of Th2 CD4 lymphocytes and polarization to a Th2 cytokine profile .88, 108, 109 Acute GVHD is thought to be a Th1 mediated disease with elevation of TNF-α and IFN-γ, whereas cGVHD is considered a Th2 mediated disease.45, 119, 120 Fibrosis and chronic inflammation are the hallmarks of cGVHD, whereas necrosis in target organs is the predominant finding in aGVHD.108 In murine models, transforming growth factor beta (TGF-β) plays an important role in the development of fibrosis. CD4+ T cells home to target tissue and secrete IL-13, a Th2 cytokine that stimulates macrophages. Activated macrophages produce TGF-β, which binds to receptors on fibroblasts causing collagen synthesis and fibrosis.108 In patients with cGVHD, serum levels of TGF-β are significantly increased.121 IL-13 itself can bind to fibroblast receptors and directly stimulate collagen synthesis. IL-4 (another Th2 cytokine) similarly binds to fibroblast receptors and stimulates collagen synthesis.122 Thus, the fibrotic changes seen in cGVHD may be accounted for by a Th2 cytokine profile and by the presence of autoantibodies to PDGFR.
Acute GVHD is a strong predictor of subsequent cGVHD.123 Therefore, steps to decrease the risk of aGVHD, such as ex vivo T cell depletion in the graft and optimization of HLA match, are undertaken. In addition, minimizing toxicity of conditioning reduces tissue damage, which can later become a target for cGVHD. For example, lesions of sclerotic, cutaneous cGVHD have been known to develop after localized radiation therapy.95 This is thought to represent an isomorphic (Koebner) phenomenon. Similarly, Martires et al. showed an association between conditioning with total body irradiation and the development of sclerotic type cGVHD.94
Patients with cGVHD should practice strict sun avoidance because of immunosuppression and a propensity to develop skin cancer. The incidence of cutaneous squamous cell carcinoma and melanoma is elevated in cGVHD, particularly in males and in those who have received total body irradiation.124 Patients taking voriconazole, a systemic antifungal medication, are at especially high risk. Voriconazole is a potent photosensitizer and a strong exacerbating factor for development of squamous cell carcinoma and melanoma.125-127 Squamous cell carcinoma associated with voriconazole can be aggressive and multifocal, with high metastatic potential. 128-130
Frequent skin and mucosal examinations should be performed to detect cancer, ulcers, or new areas of sclerosis. Serial photography is helpful in this regard. Chronic GVHD patients with a history skin cancer should be examined every six months.131 The incidence of oral cancer is also increased, and examination of mucosal surfaces should be part of routine cancer surveillance.124 Mucosal and cutaneous erosions must be treated promptly to prevent adhesions and infection.131 The skin of cGVHD patients tends to be xerotic, and generous use of emollients is recommended. Treatment of chronic ocular GVHD consists of liberal and frequent use of preservative-free artificial tears.132
If sclerodermoid change or faciitis are detected, physiotherapy is advised. Physiotherapy, which includes stretching, heat, and edema management, is considered a very important component of general cGVHD management. It should be started early to prevent contracture, reduced range of motion and disability. Faciitis occurs in approximately 1% of patients and is characterized by rock-hard, subcutaneous induration. Faciitis is progressive and can lead to serious functional impairment.131 Myositis presents with muscle pain and weakness, usually in association with skin findings. Evaluation includes electromyography, determination of creatine kinase and aldolase activity, and muscle biopsy.112, 133 Cases of myocarditis with electrocardiographic abnormalities have been described.134
Patients with cGVHD are immunosuppressed, and infection is a major cause of morbidity and mortality. Prophylactic antibiotics are an important adjunctive treatment.132 Patients are at increased risk of infection by Pneumocystis jirovecii and by encapsulated organisms such as S. pneumonia, H. influenzae and N. meningitides. Those at high risk for fungal or viral infections should also be treated prophylactically with anti-fungal and anti-viral medications. Patients lose protective immunity after HSCT, and vaccination should be instituted six months post-transplant.135 However, patients and close contacts should avoid live vaccines until full immunocompetency is achieved.
Specific treatments:
Treatment of cGVHD is complex and multifaceted, and a multidisciplinary team consisting of oncologists, dermatologists, ophthalmologists, physical therapists, plastic surgeons and others with expertise in the disorder best serves the patient. For mild disease limited to skin, topical corticosteroids (TCS) and emollients alone will suffice. The lowest potency and frequency of application should be used to avoid local adverse cutaneous effects. Because immunosuppressive effects are minimal, topical therapy is particularly advantageous in patients at high risk of malignancy relapse. Topical calcineurin inhibitors (CNI) are useful for sensitive areas such as the face and groin.136, 137 For oral mucosal involvement, TCS are also the mainstay of therapy. One study showed that the combination of topical dexamethasone and topical tacrolimus was particularly effective in reducing symptoms attributable to oral cGVHD.138
Phototherapy has antiproliferative and immunomodulatory effects, and it is effective in cutaneous cGVHD. Ultraviolet A (UVA), psoralen with UVA (PUVA), and both narrow and broad band ultraviolet B (UVB) are all treatment options. Higher response rates to phototherapy have been observed in lichenoid disease as compared to sclerodermoid cGVHD.139 Although PUVA is effective, UVA and UVB without a photosensitizer have the significant advantage of avoiding psoralen-related side effects.140, 141 In addition, the risk of skin cancer in long-term treatment is lower in UVB than in PUVA.142 In a small case series, Brazzelli et al. used narrow-band UVB to treat ten children with steroid-refractory cutaneous cGVHD. Eight had a complete response with resolution of skin lesions, while the other two showed a partial response.143 Given its wide safety margin, phototherapy is an excellent choice for patients with solitary cutaneous involvement or persistent skin lesions unresponsive to systemic immunosuppression.
Systemic therapies:
If internal organs are affected, systemic treatment is required. Systemic corticosteroids alone or in combination with a CNI are first line therapy, with starting doses of prednisone at 1mg/kg/day. Some groups recommend maintaining this dose for two weeks, after which a slow taper is begun.144, 145 Approximately 50% of cGVHD patients will respond, but long-term side effects are a concern. The addition of a CNI does not necessarily reduce mortality, but it may reduce treatment-related morbidity. In a randomized trial of low-risk cGVHD patients without thrombocytopenia, combination therapy with prednisone and a CNI did not significantly reduce transplantation-related mortality compared to prednisone alone. Combination therapy did however reduce the incidence of steroid-related toxicities such as osteonecrosis.146 Patients with cGVHD at high risk for adverse corticosteroid side effects may therefore benefit from the addition of a CNI to the initial regimen.146 In another study of high-risk cGVHD patients with thrombocytopenia, the combination of a CNI and prednisone as first-line therapy conferred a distinct survival advantage.145, 147
More than one third of cGVHD patients do not respond to corticosteroid therapy.148 Steroid refractory and steroid dependent disease are typically defined by progression of cGVHD on prednisone ≥ 1mg/kg/day for 2 weeks, stable disease on ≥ 0.5mg/kg/day for 4-8 weeks or inability to taper the dose to ≤ 0.5mg/kg/day. If a patient cannot be weaned from corticosteroids after three months, adjuvant therapies need to be considered; however, there is no established second-line treatment.91
Inhibitors of the mammalian target of rapamycin (mTOR), sirolimus and everolimus, block response to interleukin-2 (IL-2) and prevent activation and proliferation of T and B lymphocytes. In addition, they may promote the generation of Treg cells.149 Treatments of cGVHD with the combination of an mTOR inhibitor and prednisone have shown response rates of 60-70%.150, 151 This combination is well tolerated and effective for patients with sclerodermatous cGVHD.151 However, the addition of an mTOR inhibitor to a CNI appears to increase the risk of thrombotic microangiopathy.151, 152
Extracorporeal photophoresis (ECP) is a procedure wherein peripheral blood mononuclear cells are collected from the patient, treated with 8-methoxypsoralen, irradiated with UVA, and then reinfused back into the patient.153 The net result is that ECP causes apoptosis of donor effector lymphocytes while increasing donor Treg cells.154 ECP is considered an immunomodulating treatment, not an immunosuppressive treatment.155 It is effective for some cases of steroid dependent or refectory cGVHD, and one study showed lower non-relapse mortality in ECP responders.156 Another study showed that most patients were able to achieve 50% improvement in cutaneous cGVHD lesions with bimonthly ECP sessions, and over 75% of the patients were able to reduce doses of systemic immunosuppressive medication after six months.157 Sclerodermatous lesions responded better lichenoid lesions. Given its excellent safety profile, ECP is a valuable adjunctive therapy.
Numerous other medications have been used in the treatment of steroid-refractory cGVHD. These include mycophenolate mofetil158 149, methotrexate159, 160, azathioprine161, 162, pentostatin, rituximab and imatinib. Mycophenolate may be associated with a risk of thrombocytopenia, recurrent malignancy, infection and gastrointestinal discomfort.163, 164 Methotrexate is a reasonable option. It is effective, and side effects are predictable and manageable.159, 160 Azathioprine is not often used because one study showed increased nonrelapse mortality when azathioprine plus prednisone was compared to prednisone alone. 161 Pentostatin inhibits adenosine deaminase resulting in apoptosis of dividing lymphocytes, and two phase II trials demonstrated response rates of about 50%.149, 165 Side effects of pentostatin, however, are severe: serious infection, leukoencephalopathy, anemia and gastrointestinal symptoms caused a 25% drop out rate in the trials. Rituximab has shown benefit in the treatment of cGVHD, and it may also be helpful in prevention.90, 108, 166 A meta-analysis of 111 patients with cGVHD showed a combined response rate that approached 70%.167 The most notable responses were seen with the cutaneous and musculocutaneous manifestations.168 Imatinib mesylate is a tyrosine kinase inhibitor with activity against BCR-ABL-positive malignancies. It is also an inhibitor of PDGF and TGF-β signaling pathways. Several case reports and small series have shown usefulness in sclerodermatous cGVHD. 149, 165, 169,170-172
EMERGING THERAPIES
Interleukin-2 (IL-2) is a cytokine necessary for the development, expansion and function of T cells, including Treg cells, which suppress autoreactivity and maintain tolerance to self-antigens. Treg cells are reduced in chronic GVHD and other autoimmune diseases.109, 110 Koreth et al treated 29 patients with glucocorticoid refractory cGVHD with daily low-dose subcutaneous IL-2.91 Of 23 patients available for analysis, 12 had major responses in several sites, including skin, and 3 patients had partial responses. All treated patients experienced a marked increase in Treg cells compared to baseline values. None of the patients experienced recurrent malignancy, and several who continued this treatment were able to lower their glucocorticoid dose. The most common adverse effect was a dose-dependent flu-like syndrome. Serious adverse effects included renal dysfunction, thrombocytopenia, thrombotic microangiopathy, and infection. Daily low-dose IL-2 may prove to be an effective treatment, but safety and adverse side effects remain a concern.91
Mesenchymal stem cells (MSCs) also show potential value for treatment of both acute and chronic GVHD. MSCs are pluripotent bone marrow progenitor cells capable of developing into multiple mesenchymal cell lineages including osteoblasts, chondrocytes, adipocytes, cardiomyocytes, neurons, and endothelial cells.173 They can repair various tissues by homing to damaged sites and differentiating into cells of that tissue.174 They are also immunomodulators, and they inhibit T cell responses to create an immunosuppressive local microenvironment.175, 176 MSCs inhibit B lymphocytes, NK cells, and dendritic cells, but they increase Treg cell numbers.176 MSCs are obtained from bone marrow, umbilical cord blood, adipose tissue, and muscle. They exhibit immunomodulatory effects while maintaining low immunogenicity. Several studies have shown that they are safe to infuse intravenously, and HLA-compatibility between MSC donor and recipient does not appear to be important. MSCs are hypoimmunogenic, and they avoid allorecognition.
Investigations have been undertaken to determine whether MSCs can prevent GVHD. In an open-label, randomized clinical trial, Ning et al. co-administered an allograft and MSCs at the time of HSCT.177 Although co-administration decreased the incidence of both acute and chronic GVHD, it increased the risk of primary disease relapse.177 However, other investigators suggest that co-adminstation of MSCs and allograft during HSCT is safe and may actually speed the engraftment process178, 179
MSCs are also being explored for treatment of established cGVHD. Weng et al. reported good response in 14 of 19 steroid-refractory cGVHD patients treated with MSCs. Four patients achieved a complete response and 10 a partial response.148 Moreover, 9 of the 14 patients were able to reduce or discontinue immunosuppressive therapy completely, and the two-year survival approached 80%. Likewise, Zhou et al. treated 4 patients with sclerodermatous cGVHD by direct injection of MSCs into bone marrow. Following the injection, Th1 cells increased and Th2 cells decreased in all 4 patients. The reversal of Th1/Th2 ratio was accompanied by significant improvement in skin lesions and joint mobility. None of the patients showed evidence of cancer recurrence after 14 months.180 Although MSCs appear promising, more clinical trials are needed to assess effectiveness and safety.
CONCLUSION
Chronic GVHD is a major source of disability and impairment following HSCT, and it can have a profoundly negative impact on quality of life. Fibrosis is a hallmark feature and a significant cause of impairment. The immunosuppression accompanying cGVHD renders the patient susceptible to infection and cancer, the main causes of death in this condition. The pathogenesis of cGVHD is being elucidated, and promising new treatments are on the horizon.